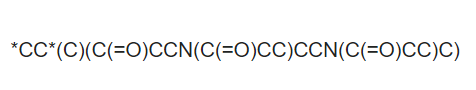I was trying to make a brunched polymer with smiles notation of the monomer like this:
I got the error of missing rules.
How can I add the missing rule? What should I add in the .define file? Do I add the resulting smiles with terminator-1 monomer-terminator or just the monomer? I could not find any document on this.
I also tried run the convert.sh script with the original .define file, but it stuck in the “converting” state for two days.
Your idea of using the .define files is a correct one. I find it somewhat puzzling though, that running convert.sh hangs. It sounds like the code is waiting for input. Normally it runs through in less than a second creating opls-ua.top, opls-ua.prm, opls-aa.top, and opls-aa.prm.
The define file holds chemical rules and any additional parameters with respect to the provided src/oplsaa.{par|sb}. Define files allow for different paragraphs identified with the keyword ITEM. Each paragraph ends with an ITEM END, not unlike the syntax used by EMC Setup. ITEM MASSES (will be ITEM MASS in future) and ITEM RULES form required paragraphs, others are optional.
Chemical rules refer to index or type numbers as given in the first column of src/oplsaa.par, e.g. index 54 refers to a united-atom fluor and index 140 to hydrogen connected to an sp3 carbon. Extra types are created in ITEM EXTRA, where new indices refer to old ones through separation with a decimal dot, e.g. index 140.1 represents a new type based on index 140. These new or extra types are used in the ITEM RULES paragraph.
The ITEM RULES paragraph defines the typing rules by means of chemical environment. E.g. index 138 refers to an sp3 carbon in methane for which the rule is
138 C(H)(H)(H)(H)
which represents a carbon to which four hydrogens are connected. The concept of SMILES are used to describe chemical environments. The first atom represents the atom for which the type is defined – a carbon in the above example. Following atoms in a SMILES represent the connected atoms. In total, a tree can be defined thus describing the chemical environment of the atom to be typed.
Sometimes bonded interactions are missing. Additional information can be provided in the ITEM BOND, ITEM ANGLE, etc. paragraphs.
No, normally executing convert.sh in the directory it appears should do the trick, provided emc_opls.pl is available in your PATH. You can put some print statements in convert.sh to see how far a script is getting while being executed.