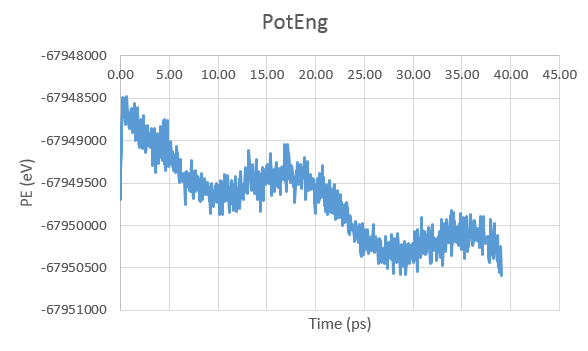
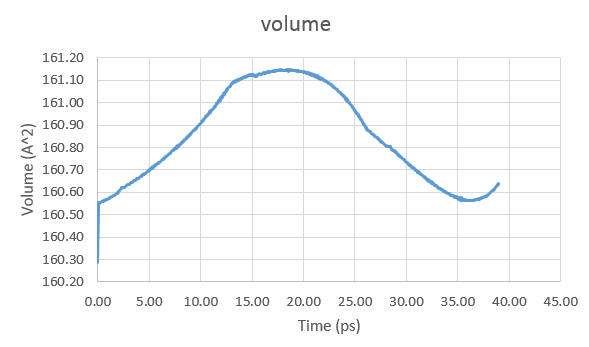
I am running a simulation of silicon atoms, first in NPT ensemble and the
pressure fluctuations are very low (plus or minus 20 bars) in my 14mil atom
simulation, which is great. But when I switch from NPT ensemble to NVE, I
get fairly large pressure fluctuations (for instance right now the pressure
is at ~800 bars). I believe it will swing back down the other way as well.
that doesn't sound like a fluctuation. perhaps, your volume for NVE
was by chance significantly away from the equilibrium volume.
Are there any measures that can be taken to reduce this? I understand that
please keep in mind, that different materials have a different
compressibility leading to either larger or lesser changes in pressure
due to a given change in volume.
when you switch from NPT to NVE, you don't want to switch to the
*instantaneous* box dimensions, but rather sufficiently averaged ones.
there are other factors, that have an impact as well.
with the NVE ensemble, I am fixing the volume and energy and thus pressure
fluctuates, but I would not except such a huge pressure wave in the system
under equilibration. Is this sort of thing normal to see, or is there a
procedure for reducing these fluctuations?
in a properly tuned and equilibrated NPT simulation, the switch to NVE
should not incur significantly larger pressure fluctuations. the time
constant for adjusting the volume with fix npt should be
*significantly* larger than typical pressure fluctuations, or else the
system will couple to strongly to the fictitious DOFs of the
thermostats, and unphysical things may happen. if you use a "drag"
factor to artificially suppress too strong reactions to pressure
fluctuations, then the switch to NVE will simply demonstrate, how much
you have impacted the simulation before.
it is difficult to make more specific statements without knowing more
details. please keep in mind, that you get out what you put in, since
computer software doesn't know anything about "reasonable". it simply
blindly does exactly what you tell it, even if that is not what you
mean.
axel.