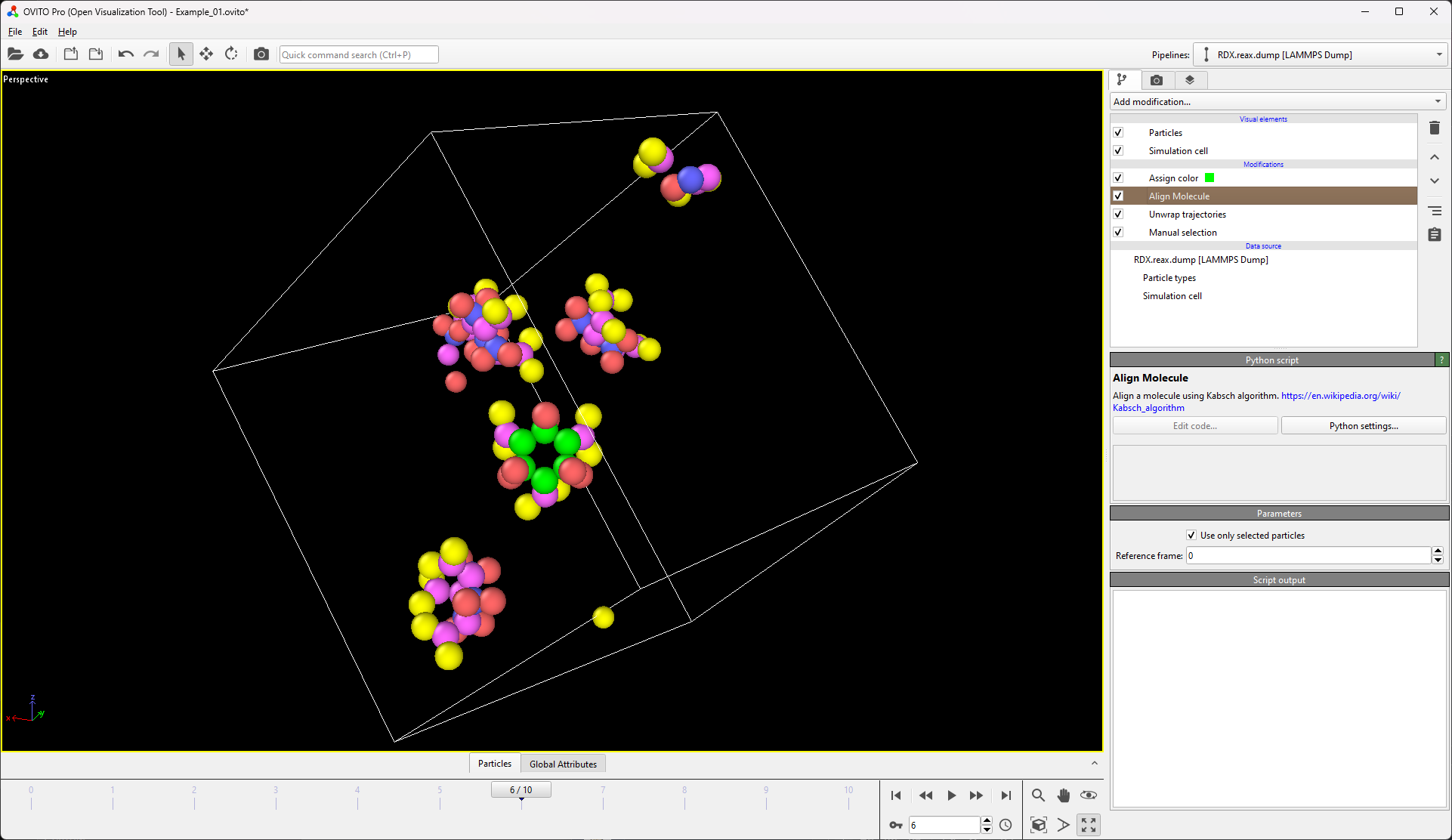
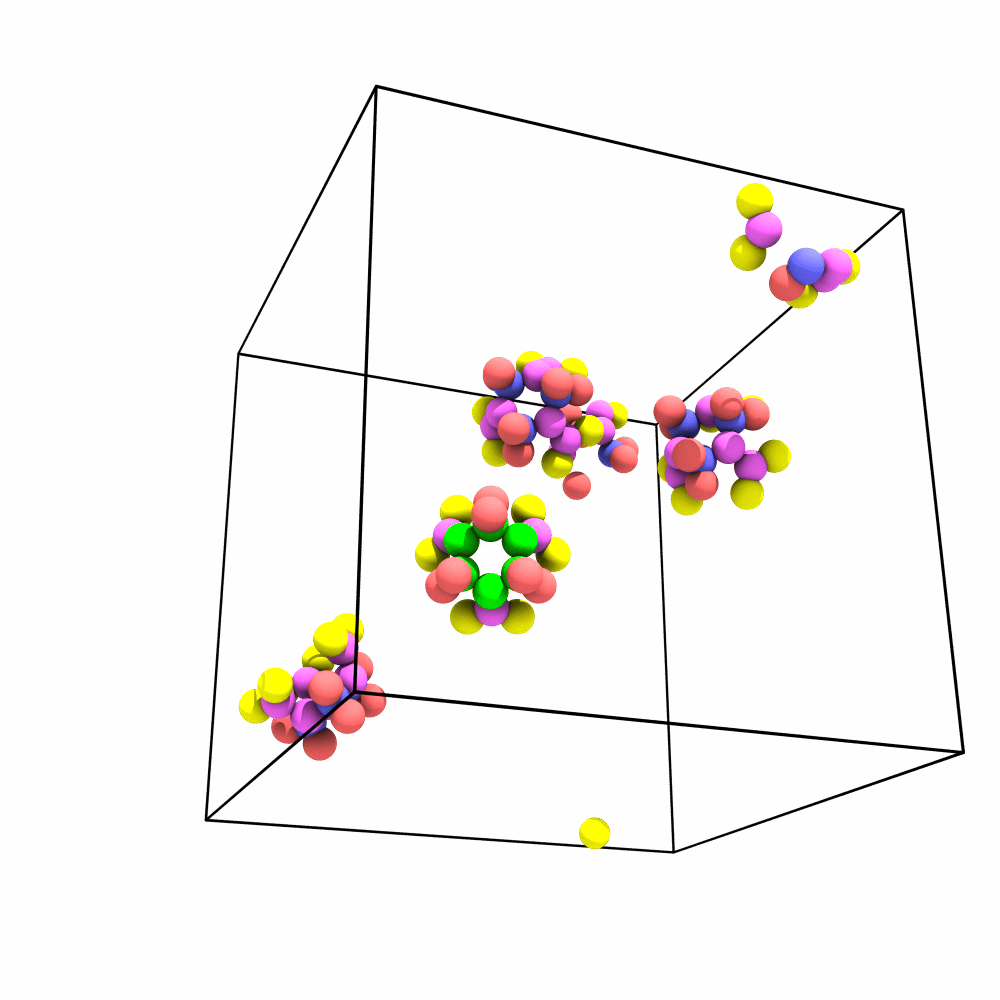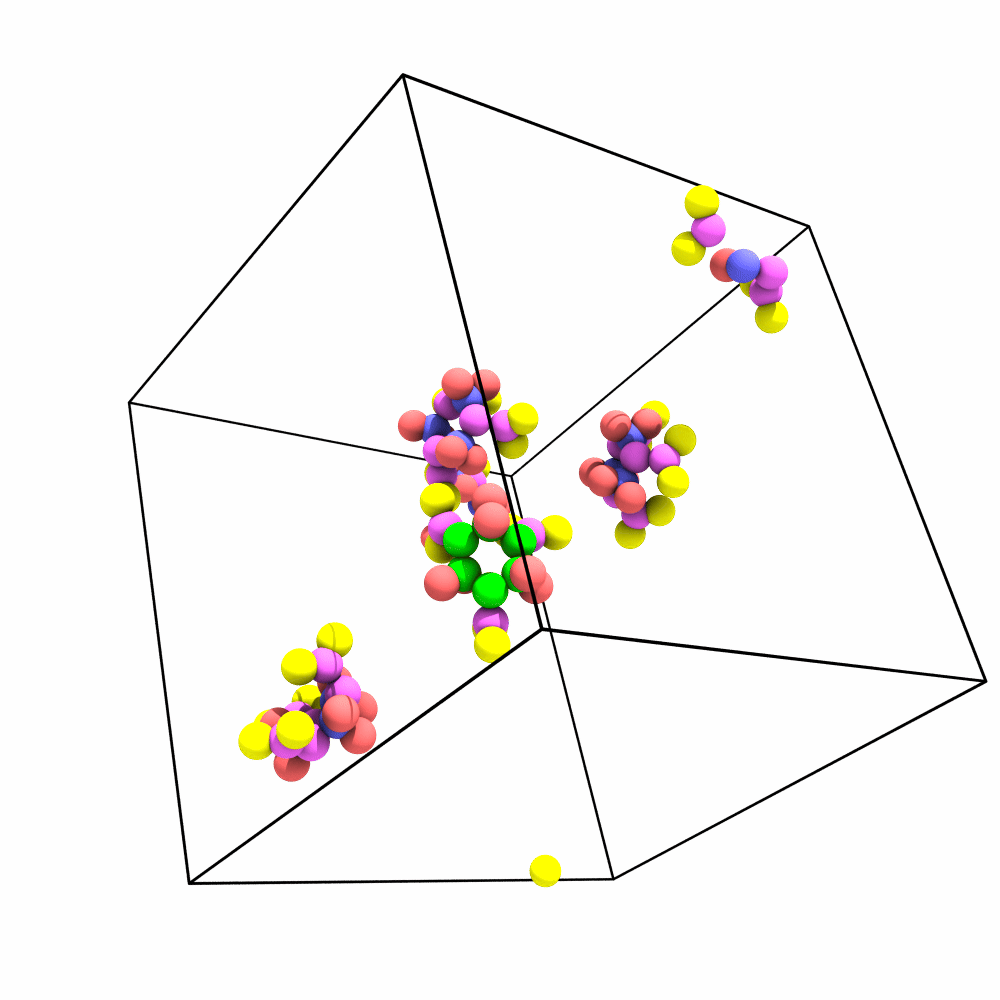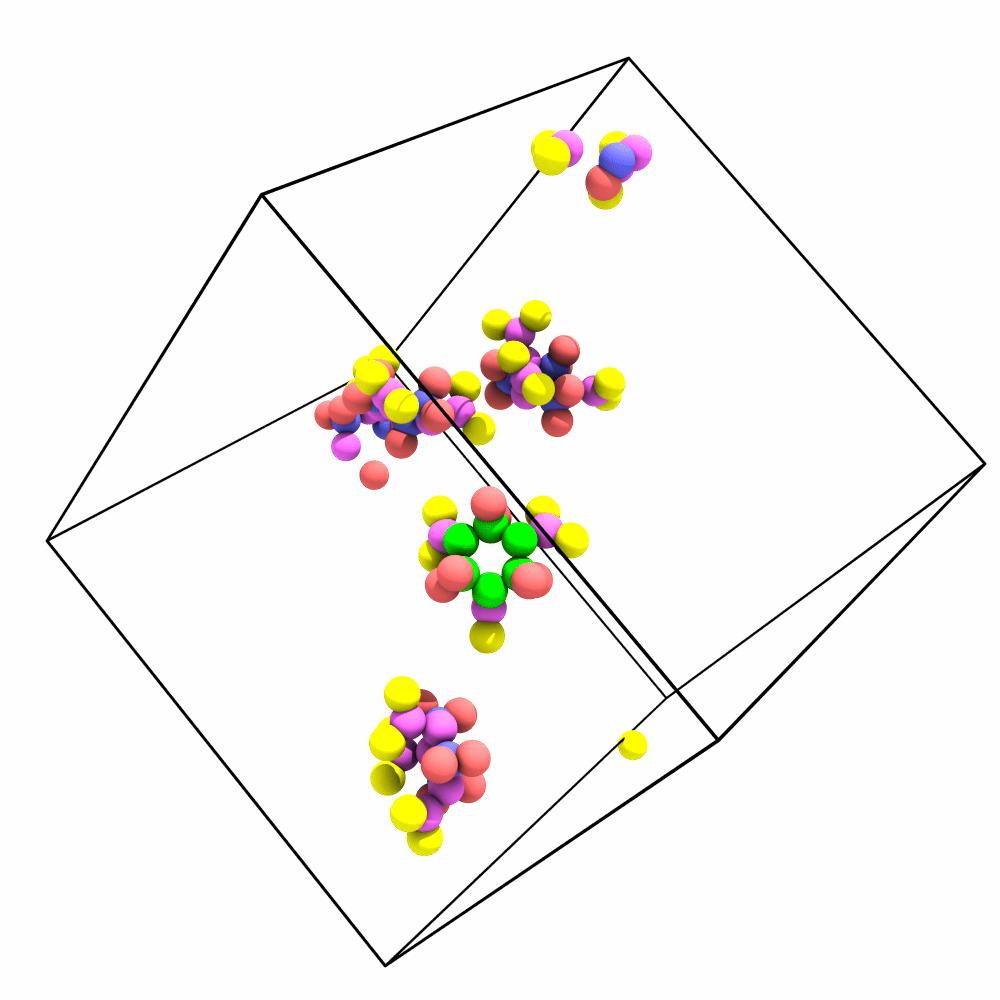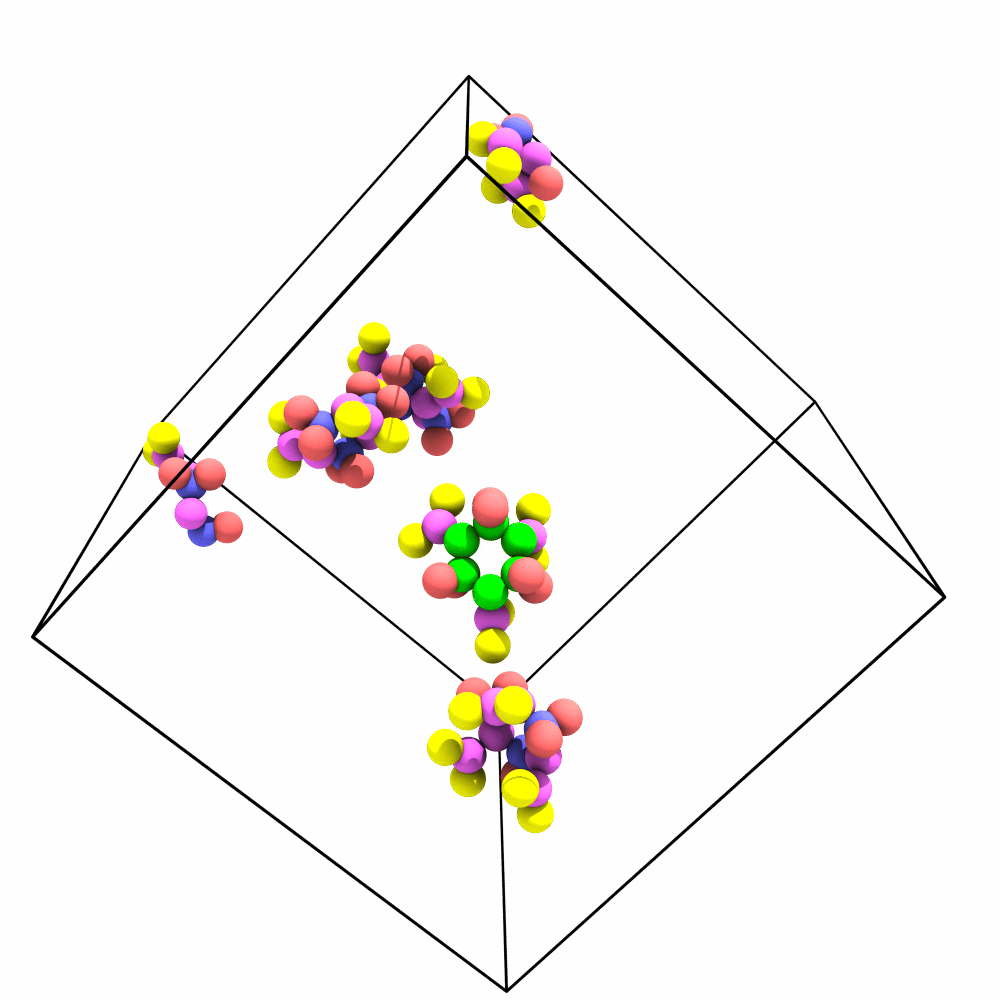Is there a way of imaging/aligning the trajectory on a set of atoms for visualization? I have a system that has a lot of rotation and translation so it’s hard to produce a movie that depicts the internal changes of the system.
Apologies if this question is already replied somewhere else, I have been looking around the docs and here and couldn’t find it.
Thank you for your question. OVITO does not currently offer a built-in function to accomplish this - apart from more complex solutions involving the Cluster Analysis modifier for dynamic calculation of the center of mass and a user-defined Python modifier function.
However, the OVITO team would like to integrate such a function into a future version of the software. Since we have a different scientific background, we lack a good overview of the typical requirements that such a feature should fulfill in practice to be most useful. You could support us by
providing a more formal definition of the desired feature. How should this function work (technically and from a user perspective)? What would the user workflow ideally look like?
giving us access to your molecular trajectory so that we have a suitable test case.
I would be happy to discuss the above questions with you and other people in this forum.
The way me/my lab would benefit from this would be to be able to make a selection (i.e. Particle Type 1) and then align the trajectory with respect to those particles in frame 1. A tool that does this is present in the vmd gui (RMSD Trajectory Tool) in case that helps with reference. The idea would be to minimize RMSD, by applying only translations and rotations for each frame. The tool could in turn then output the RMSD of the system after doing such alignment (since that has to be computed either way).
I’m not sure how feasible the implementation of that is, but happy to discuss further with you/the team/the community.
I’m happy to share a small trajectory with you if that helps - I don’t seem to be able to attach files in the forum.
Thank you for these suggestions. You are welcome to send us your trajectory by email to [email protected].
Next, we will collect a few detailed questions in an internal discussion, e.g. how to handle periodic boundary conditions correctly and what to do if the selection of atoms comprises more than one molecule (does bond topology even play a role?). I’ll get back to you here.
I tried this, and it works amazingly well for spherical particles! However, when I use ellipsoids, they behave oddly.
Relvant Information:
The visualization is from a dump file generated by LAMMPS. To view it correctly (whether using this package or not), it’s necessary to adjust the column mapping and divide the AsphericalShape values by 2.
Thank you very much for your feedback. Could you please provide an example trajectory and an OVITO state file that illustrate the problematic behavior? I would like to investigate this further. If you’re not comfortable sharing these files publicly, please feel free to send me a DM or email.
The issue occurred because the affine transformation modifier did not affect the orientation property of your particles. This short coming has been resolved in the latest development build of OVITO, which you can download from our development builds page.
Please note that, due to technical difficulties, a macOS development build is currently unavailable.