Hi. First post here. I’m a student at Bunker Hill Community College and have been working on a script to convert the coordinate and topology files produced by AmberTools, respectively .inpcrd and .prmtop, into LAMMPS input using read_data.
I’d like to give a short description of the workflow in development. At current I’ve got something which can relatively painlessly turn many nicely-formatted .pdb files into LAMMPS input. Most-all of these steps are made easy on my machine with bash scripts rather than direct terminal input.
- Obtain .pdb file or other source of atom coords. If necessary, rename atoms or residues to align with the atom names used in AmberTools’s .dat and .lib force field parameter files, take any other steps needed to ensure data will be legible for teLeap in the next step.
- Start teLeap, load force fields appropriate for your .pdb file. Ensure parameters were assigned correctly. Add a bounding box. Save .prmtop and .inpcrd files named how you want.
- Run converter script in terminal with filenames as command-line arguments. Using Python library parmed, all atoms, bonds, angles, dihedrals, lj types are read from the .prmtop and .inpcrd files and (hopefully) faithfully converted to read_data’s format before being saved to a new file.
- Run lammps input script. Use name of file saved from conversion as a CLI argument. Thermo and picture output is saved.
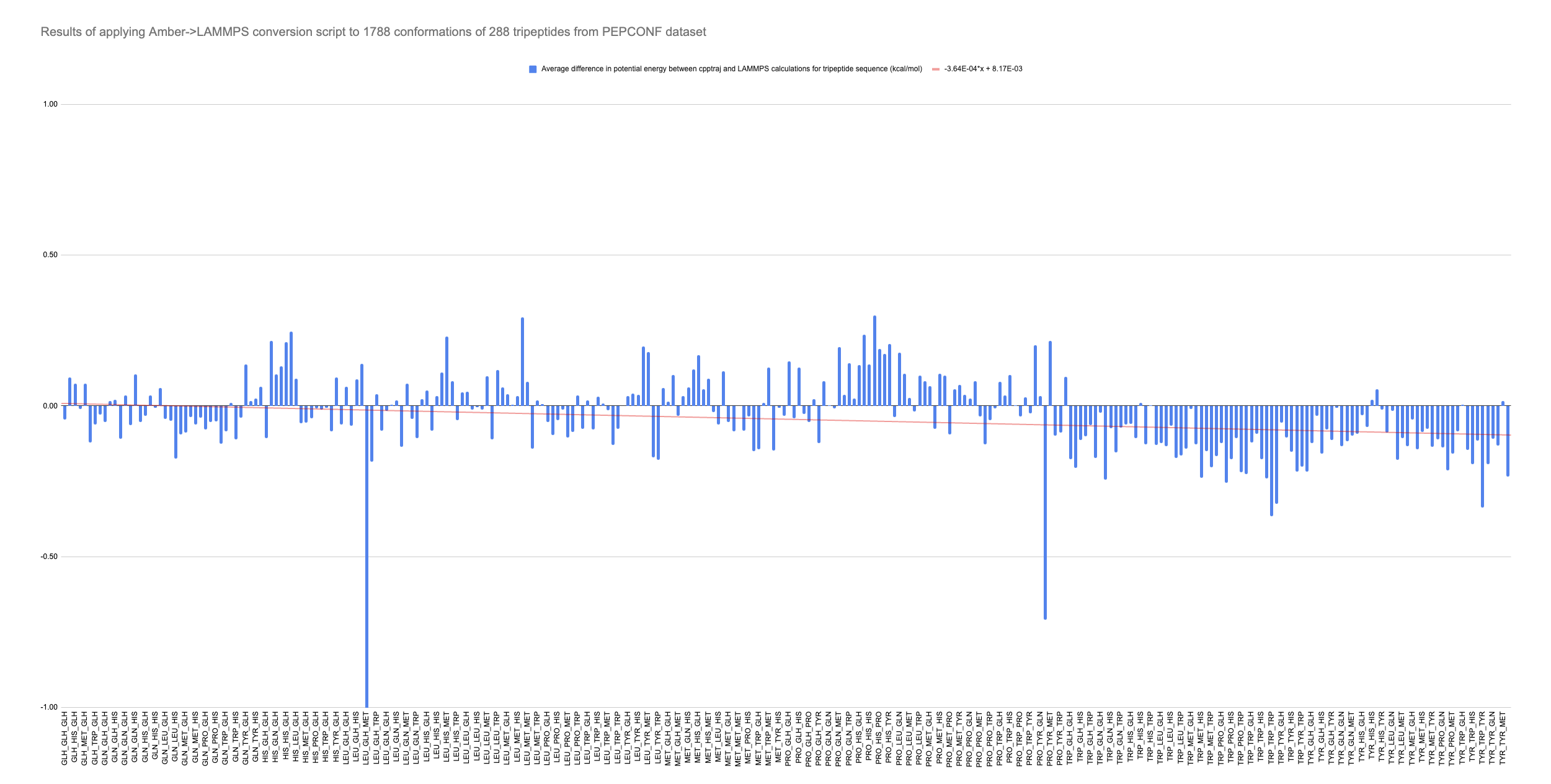
Hoping to verify conversion worked, I generated over 1000 pdb files from tripeptide conformations in the PEPCONF dataset with another script. Using these, I fed them through teLeap, then used my converter to generate many LAMMPS data files, which I ran with run 0 to ensure that thermo data would written and saved quickly. I then followed a similar procedure to get potential energy from each conformation using AmberTools’ cpptraj. All energy values were saved to a .csv file for analysis.
Fortunately, the average result for the LAMMPS calculation was less than 1% off the corresponding cpptraj number. Forcefield params are accurately translated.
From here, I intend to try some more dynamic and sophisticated methods of validation involving solvation, more residues, ligands, etc. A longer-term objective is to build a protein-ligand complex in teLeap and then use LAMMPS simulations to analyze binding affinity.