There is not enough information here to make any specific assessment.
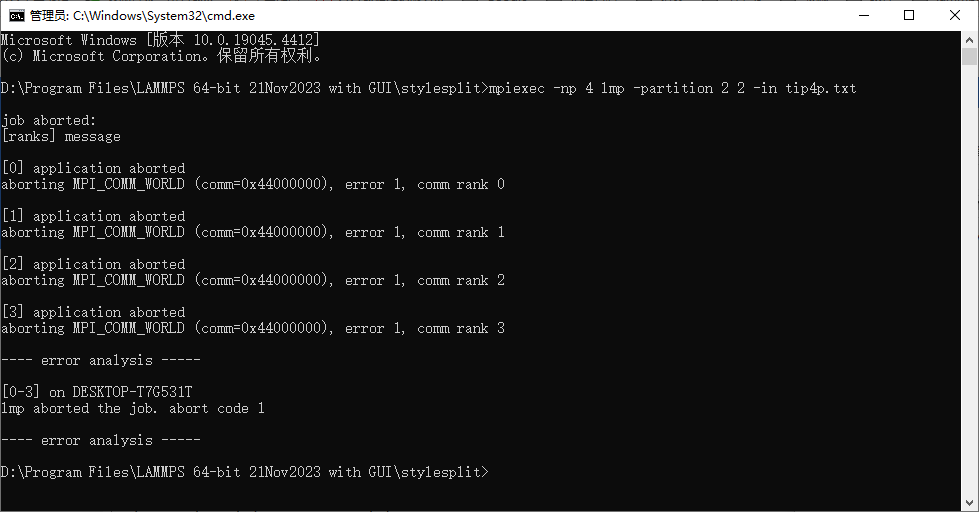
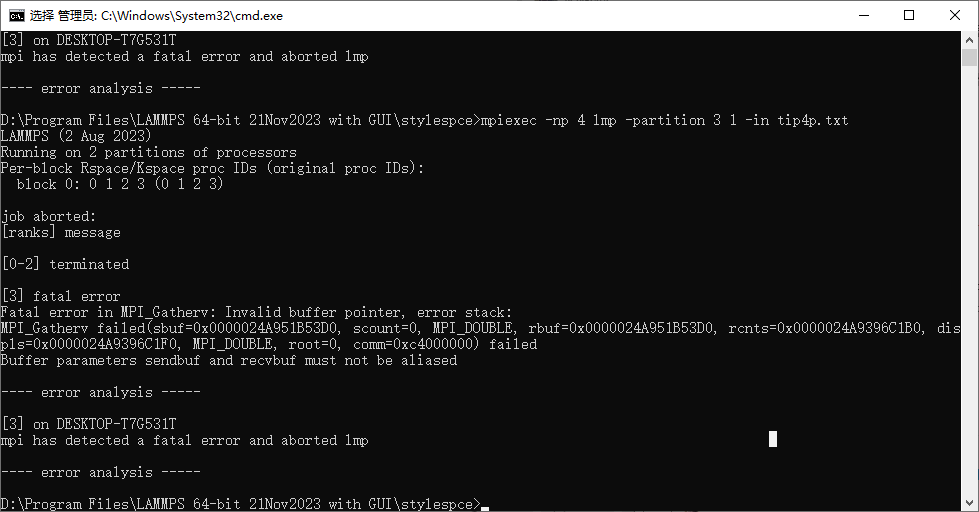
You should provide the complete input deck to be able to reproduce your simulation. There must be an error in it that is not shown to the screen. Have you checked the screen.? files??
Also you should upload the “MPI task timing breakdown:” information for your regular verlet run.
As a point of reference, here is the timing output for the “rhodo” benchmark example. First for regular verlet with for MPI tasks:
Loop time of 6.64045 on 4 procs for 100 steps with 32000 atoms
Performance: 0.651 ns/day, 36.891 hours/ns, 15.059 timesteps/s, 481.895 katom-step/s
99.6% CPU use with 4 MPI tasks x 1 OpenMP threads
MPI task timing breakdown:
Section | min time | avg time | max time |%varavg| %total
---------------------------------------------------------------
Pair | 5.0777 | 5.1938 | 5.3921 | 5.2 | 78.22
Bond | 0.19833 | 0.20452 | 0.21836 | 1.8 | 3.08
Kspace | 0.60426 | 0.79964 | 0.92044 | 13.2 | 12.04
Neigh | 0.16659 | 0.16662 | 0.16664 | 0.0 | 2.51
Comm | 0.053485 | 0.053931 | 0.054229 | 0.1 | 0.81
Output | 0.00019315 | 0.00020414 | 0.00023533 | 0.0 | 0.00
Modify | 0.20754 | 0.20911 | 0.21193 | 0.4 | 3.15
Other | | 0.0126 | | | 0.19
and here for using verlet/split partition 0:
Loop time of 7.5521 on 3 procs for 100 steps with 32000 atoms
Performance: 0.572 ns/day, 41.956 hours/ns, 13.241 timesteps/s, 423.723 katom-step/s
99.7% CPU use with 3 MPI tasks x 1 OpenMP threads
MPI task timing breakdown:
Section | min time | avg time | max time |%varavg| %total
---------------------------------------------------------------
Pair | 6.5852 | 6.6661 | 6.7238 | 2.3 | 88.27
Bond | 0.24862 | 0.26474 | 0.28059 | 2.5 | 3.51
Kspace | 0 | 0 | 0 | 0.0 | 0.00
Neigh | 0.21704 | 0.21707 | 0.21711 | 0.0 | 2.87
Comm | 0.050077 | 0.10879 | 0.20697 | 21.2 | 1.44
Output | 0.0002199 | 0.00022339 | 0.00023007 | 0.0 | 0.00
Modify | 0.13303 | 0.13553 | 0.13763 | 0.5 | 1.79
Other | | 0.1597 | | | 2.11
and partition 1:
Loop time of 7.55064 on 1 procs for 100 steps with 32000 atoms
Performance: 0.572 ns/day, 41.948 hours/ns, 13.244 timesteps/s, 423.805 katom-step/s
95.9% CPU use with 1 MPI tasks x 1 OpenMP threads
MPI task timing breakdown:
Section | min time | avg time | max time |%varavg| %total
---------------------------------------------------------------
Pair | 0 | 0 | 0 | 0.0 | 0.00
Bond | 0 | 0 | 0 | 0.0 | 0.00
Kspace | 2.1499 | 2.1499 | 2.1499 | 0.0 | 28.47
Neigh | 0 | 0 | 0 | 0.0 | 0.00
Comm | 0 | 0 | 0 | 0.0 | 0.00
Output | 0 | 0 | 0 | 0.0 | 0.00
Modify | 0 | 0 | 0 | 0.0 | 0.00
Other | | 5.401 | | | 71.53
There is only 12% of the total time spent on Kspace, so using a 3 1 partition split will allocate 25% of the resources, i.e. more than double to the Kspace partition and it will run slower since it is not in parallel across 4 processors plus there is overhead from verlet/split.
The result shows that regular MPI runs are over 10% faster (6.64 seconds versus 7.55 seconds).