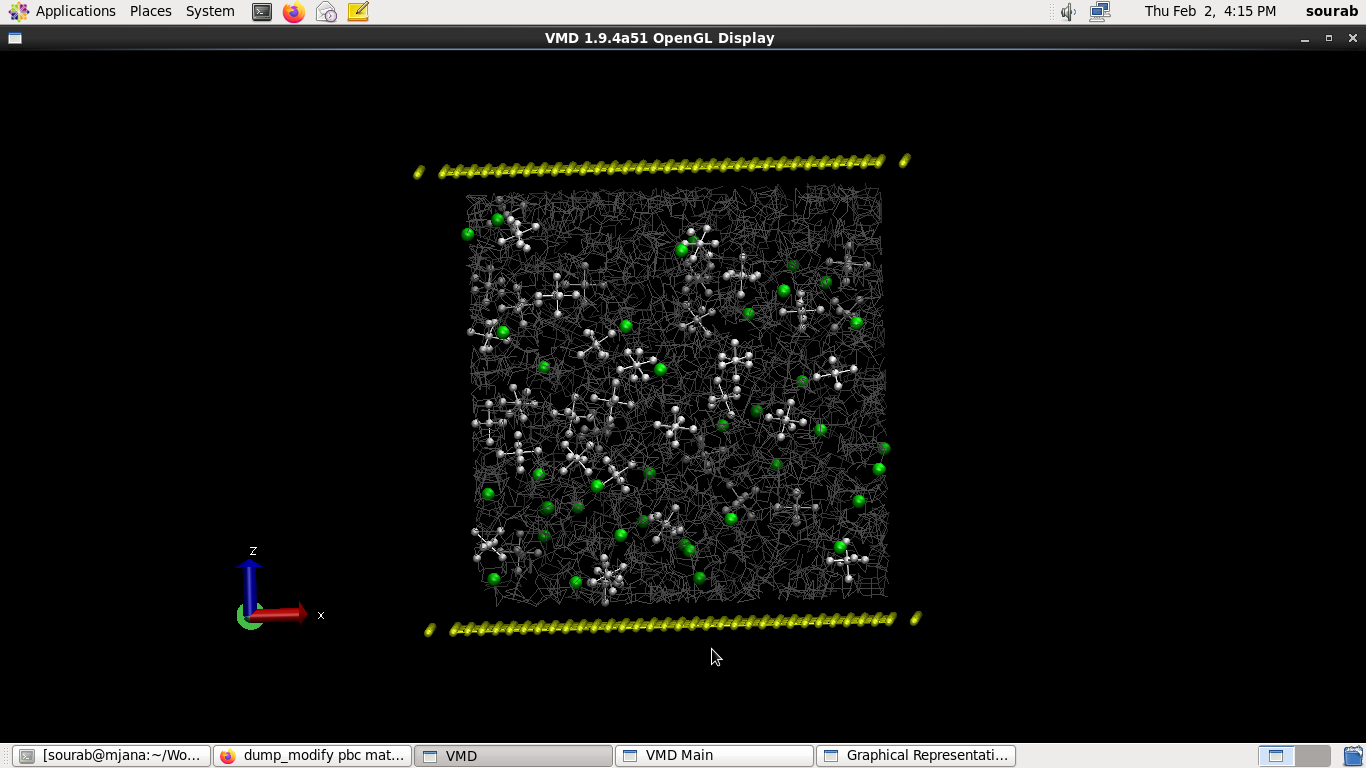
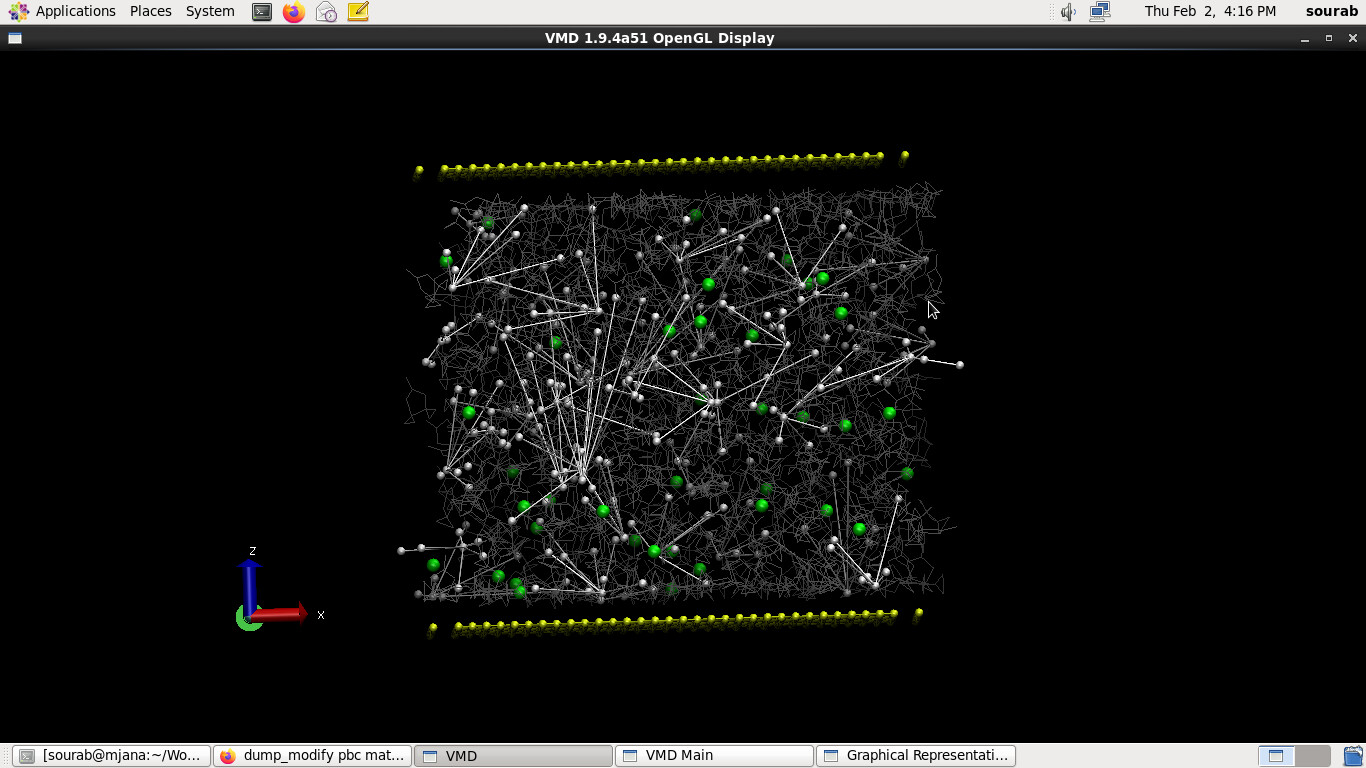
I’m using LAMMPS version 23 Jun 2022, on Centos 6. I have installed the reaxFF package bundled with lammps and ECHEMDID-23jun2022
Hello, I’m trying to simulate the behavior of 1M of LiPF6 in Ethylene Carbonate under an electric field in order to establish a baseline for my future simulations. In this process I decided to use ReaxFF and ECHEMDID, as the force field and electric field implementations to run my simulations. However, upon equilibration, my PF6 molecule splits apart with all the flourine atoms repelled from the P center.
I would like assistance from people who have conducted similar simulations and whether this kind of behaviour has been observed earlier.
Here is a relevant snippet from my input deck
#################################################
#init
units real
atom_style charge
boundary p p s
read_data electrolyte_LiPF6_40A.data
pair_style reaxff lmp_control
pair_coeff * * ffield.reax.CHOSLiPF C C C F F F F F F H H H H Li O O O P
#minimisation
fix chg all qeq/shielded 1 10 1.0e-10 500 reaxff
thermo 1
minimize 1.0e-12 1.0e-12 10000 10000
#ECHEMDID
variable zmin1 equal bound(all,zmin)
variable zmax1 equal ${zmin1}+0.2
variable zmax2 equal bound(all,zmax)
variable zmin2 equal ${zmax2}-0.2
fix 0 all property/atom d_locpot d_lap
compute 0 all property/atom d_locpot d_lap
region bound1 block INF INF INF INF ${zmin1} ${zmax1} units box
group z_left region bound1
region bound2 block INF INF INF INF ${zmin2} ${zmax2} units box
group z_right region bound2
set region bound1 d_locpot 0.0
set region bound2 d_locpot 0.0
fix 2 all echemdid 1 k 6.0 rc 4.0 norm 0.629967 nelec 25 z_left z_right volt 10.0
timestep 0.1
fix tres all nvt temp 1 300 100
run 30000
unfix tres
fix mynvt all nvt temp 300 300 100
run 50000
write_data electrolyte_min.data
################################################
A straightforward guess is that the methodology has reacted poorly to such a strong electric field / large potential difference, mistakenly polarising the phosphorus from the fluorines in PF6 along the intended applied field, resulting in the topologies you’re seeing.
As to why that is happening, I don’t know. Could be any or all of: reaxff+echemdid simply won’t work for this system; your fix qeq needs to come after echemdid, not before; reaxff won’t work with this system because it doesn’t account for long range electrostatics; etc. etc.
A quick search shows that there are a few groups around the world who specialise in this technique – if you have more advanced questions, you may have to ask them directly.
Rather than simulating a big complex system, I suggest you run simulation of a small number of individual components (E.g. \textsf{PF}_6) by themselves and validate whether your force field and geometry is adequate. After that, I would try to set up a more complex geometry but still without the ECHEMDID add-on and see if that gives meaningful results, and only if that is successful, too, turn on the add-on feature. That way it is much easier to determine where the issue is (force field, geometry, combined structure, ECHEMDID add-on).
In fact, it is not possible to “fix all echemdid”, first of all the problem of the charge calculation that will lead to the electrolyte, and echemdid should only be applied to the electrode part. Secondly, because the potential is “diffused” from both sides in echemdid, when diffused to the middle, the calculation of the atom d_locpot in the middle part will be abnormal, resulting in the collapse of the simulation. I hope you found this helpful.
The ECHEMDID command specifically describes regions where the fix will be applied, so I’m not sure the “fix all echemdid” will cause an issue? Even the examples provided by the makers of the plugin seem to be using fix all (however their system only contains two types of atoms and no electrolytes).
Would applying ECHEMDID only to my electrode groups yield better results?
If you just want a result that looks normal and stable, let the potential spread from the electrode on one side and the solid-liquid distance on the other side be greater than the cut-off distance you have set (4 angstroms). Based on my attempts with similar models, there really doesn’t seem to be an obvious error. But the truth is that, from a scientific point of view, echemdid is the right thing to be ‘fix’ at the electrode, depending on the charge changes at the electrode to influence the structural fluctuations of the electrolyte.
In addition, it seems that you did not do enough research on echemdid. The example provided by onifrio is only an example and certainly misled me at first. Try searching for ‘hacortesp’ on github or searching for echemdid.There are complete examples in many places. Finally, please don’t take my answer completely at face value. I have only been dealing with this subject for a few months and can’t claim to have a deep understanding of it. I hope you found this helpful.
Forget to add that the nvt ensemble doesn’t seem to fit very well with reaxff, at least in my opinion, thermostats often fail. You can try the nve ensemble and change the thermostat.