I am using LAMMPS version 22 Dec 2022 on a CentOS system to conduct a shear box deformation simulation. During the simulation, I used the write_restart command to output an atomic configuration at a specific moment as restart_shear.equil, and then continued the simulation for an additional 100,000 steps. Subsequently, I read the output restart_shear.equil from a different file and continued the simulation for another 100,000 steps.
I monitored the epair changes obtained using thermo_style for both 100,000-step simulations (the original one and the one reading the restart file). Initially, the two were almost identical, but after approximately 50,000 steps, differences began to emerge.
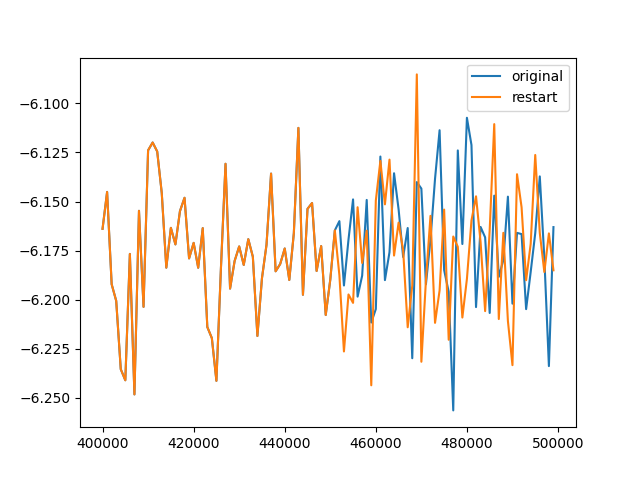
Is this a normal phenomenon due to finite computational precision, or have I made a mistake in my settings somewhere?
Attached:
Original LAMMPS input file
units lj
dimension 3
boundary p p p
atom_style atomic
lattice fcc 1.2
region box block 0 8 0 8 0 8
create_box 2 box
create_atoms 1 box
group smaller_atoms id 1:19652:5
set group smaller_atoms type 2
change_box all triclinic
mass 1 1.0
mass 2 1.0
neighbor 1.0 multi
neigh_modify delay 0
comm_modify mode multi
pair_style lj/cut 2.5
pair_coeff 1 1 1.0 1.0 2.5
pair_coeff 2 2 0.5 0.88 2.5
pair_coeff 1 2 1.5 0.8 2.5
pair_modify shift yes
velocity all create 1.0 882564 dist gaussian rot yes
timestep 0.005
fix eq all nvt temp 1.0 1.0 $(100.0*dt)
thermo 1000
thermo_style custom step temp epair etotal press vol pxy
run 200000
unfix eq
velocity all ramp vx 0 $(0.002*ly) y 0 $(ly) sum yes units box
fix 1 all deform 1 xy erate 0.002 remap v
fix 2 all nvt/sllod temp 1.0 1.0 $(100.0*dt)
run 200000
write_dump all custom dump_config_shear.colloid id type x y z
write_restart restart_shear.equil
run 100000
Restart LAMMPS input file (restart lmp file)
read_restart ./restart_shear.equil
neighbor 1.0 multi
neigh_modify delay 0
comm_modify mode multi
timestep 0.005
fix 1 all deform 1 xy erate 0.002 remap v
fix 2 all nvt/sllod temp 1.0 1.0 $(100.0*dt)
thermo 1000
thermo_style custom step temp epair etotal press vol pxy
run 100000