Dear Members of the LAMMPS Community,
I find myself facing a challenging issue and would greatly appreciate any assistance with my current LAMMPS simulation. My research involves investigating the motion of an edge dislocation under a constant shear stress. The simulation cell I am utilizing closely resembles the one depicted in the figure below:
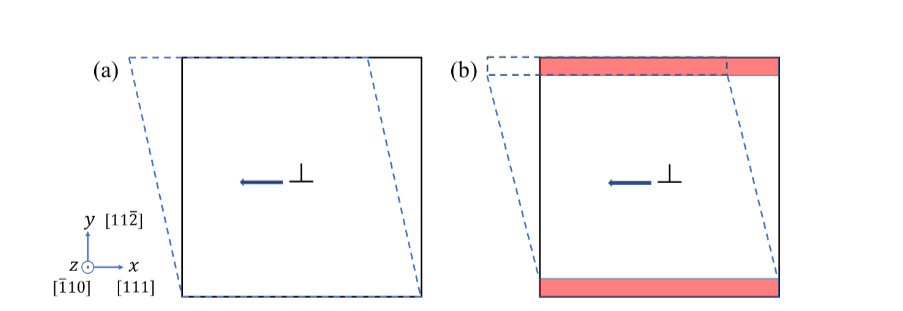
The dimensions of this cell are as follows: lx = 450A, ly = 320A, and lz = 100A.
To create the edge dislocation, I employed ATOMSK and subsequently performed an energy minimization using the conjugate gradient (CG) method. Afterward, I conducted an NVT (constant number of particles, volume, and temperature) simulation to equilibrate the structure at 0.01K.
Finally, I applied a constant stress at top red layer using the fix aveforce command and continued the simulation with fix nvt for a duration of 120ps.
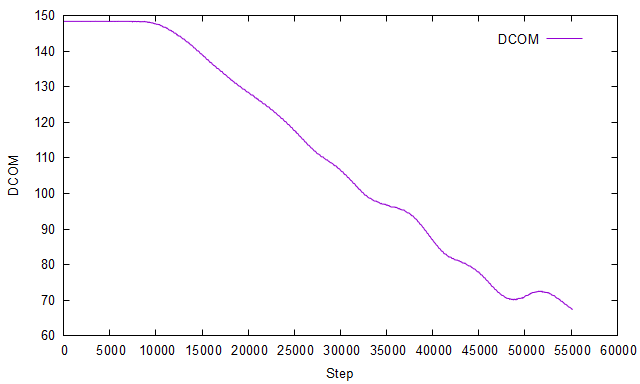
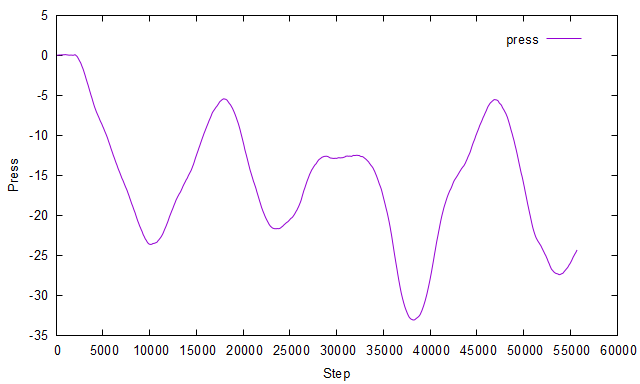
I had anticipated that the shear stress (pxy) obtained from the simulation would match the applied stress. However, contrary to my expectations, the shear stress exhibits variations with each timestep, as illustrated in the figure below:
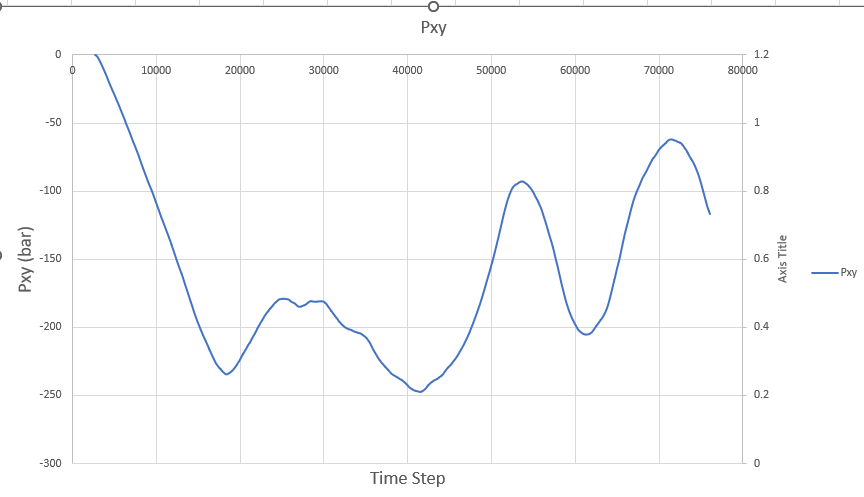
I find myself perplexed and unable to pinpoint the source of this discrepancy. Any insights or guidance you can provide would be immensely helpful in resolving this issue. One question I have is whether I should apply shear stress under the fix npt ensemble.
I have attached a snippet of my code that is used to apply stress below:
# Calculation of Forces in Upper and Lower Group of Atoms
variable natoms_upgrp equal count(upgrp)
variable natoms_logrp equal count(logrp)
variable stress equal 18 # MPa
variable lx0 equal lx # A
variable lz0 equal lz # A
variable force equal ${stress}*${lx0}*${lz0}*1e-14 # N
variable fx_up equal (${force}*1e9)/(${natoms_upgrp}*1.602)
variable fx_lo equal (${force}*1e9)/(${natoms_logrp}*1.602)
# 1eV/A = 1.602e-19/1e-10 = 1.602e-9N
# 1N = 1e9/1.602
print ${fx_up}
print ${fx_lo}
# Add force & Set force - Apply Forces on top surface
fix f4 upgrp aveforce ${fx_up} 0. 0.
fix f2 upgrp setforce NULL 0.0 NULL
fix f3 logrp setforce 0.0 0.0 NULL
fix f6 upgrp rigid group 1 upgrp
fix f1 mobile nve
fix f7 mobile temp/rescale 10 ${temperature} ${temperature} 0.03 1
# fix f1 mobile nvt temp ${temperature} ${temperature} ${tdamp}
# Variables Defination
variable f4ax atom f_f4[1]
# Outputs
dump d5 all custom 500 A5_result_s5 id x y z c_stress[*] c_cysm v_f4ax v_disid
dump d6 all custom 500 B6_result_s6 id xu yu zu c_stress[*] c_cysm v_f4ax v_disid
fix fout2 all ave/time 1 250 300 f_f4[1] file forces.txt
run 120000