lammps version 2May11, VMD 1.9
Problem: I am using VMD to visualize lammps output of THF in polymer. VMD is displaying artifactual bonds between H atoms of THF and I would like to know how to eliminate them.
To view my output in VMD, I take the following steps:
-
my lammps input file contains the following line to print coordinates info to a dump file, dumpdiff.lammpstrj:
dump dumpdiff diffusant custom 50 dumpdiff.lammpstrj id type xu yu zu vx vy vz
-
In VMD, I use the TCl extension command line to read in topology info using Axel’s topotools functionality:
topo readlammpsdata test.data
-
On TCl extension console, I use the topotools ‘guessatom’ command to inform VMD of atom types and radii, etc:
topo guessatom lammps data
-
Finally, I load the lammps trajectory data, dumpdiff.lammpstrj.
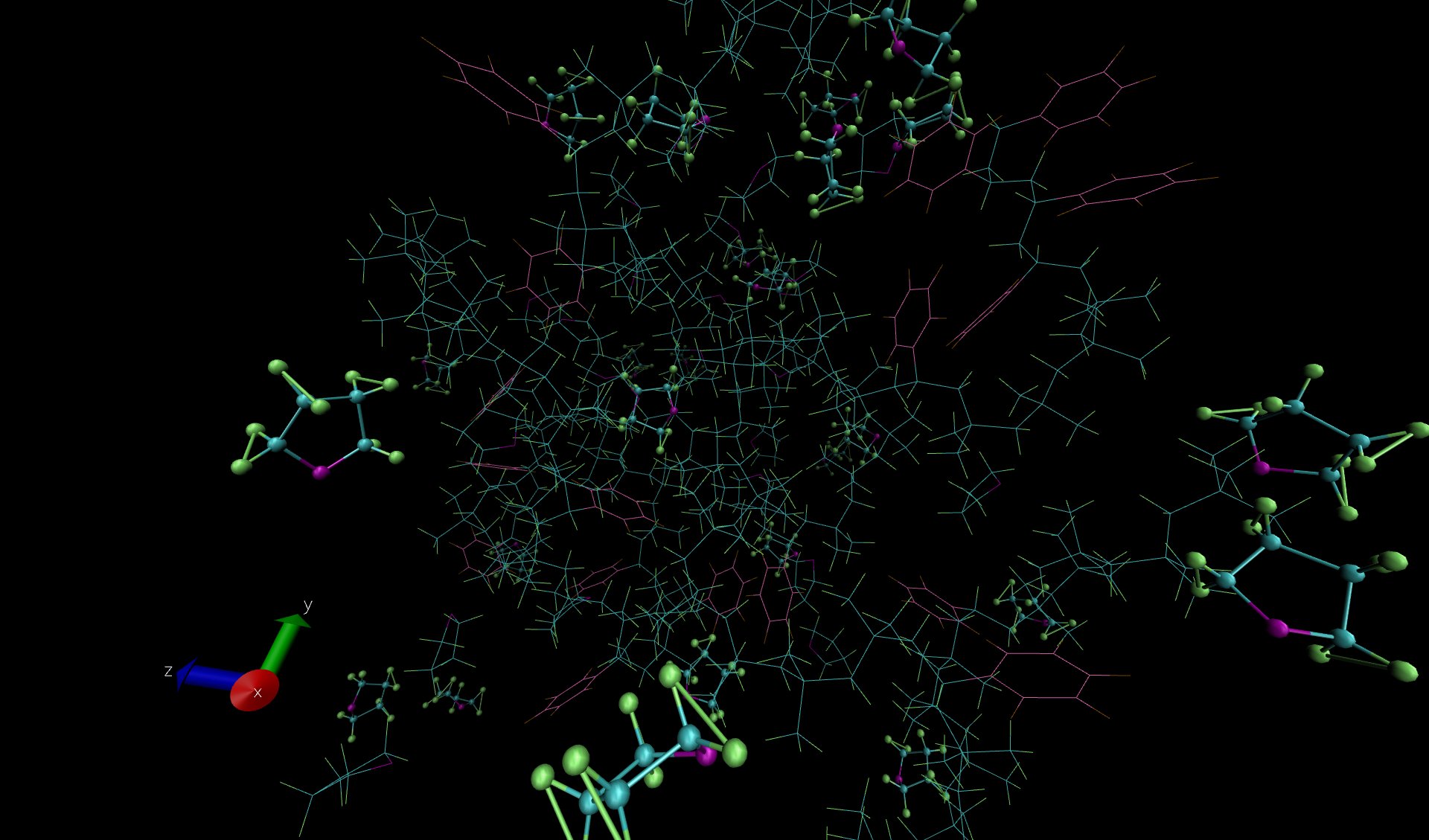
The resulting picture in VMD still shows bonds between H atoms. I checked that bonds are not (erroneously) specified in the lammps data file. I also verified that the number of bonds transmitted from the lammps data file to VMD ( topo numbonds) is the same as the number of atoms in the lammps data file. (Both have 1765 bonds). I have attached a snapshot of the visualization from VMD, as well as the lammps data file and input file.
Thanks,
Chris
test.data.gz (114 KB)
test.in (903 Bytes)
Chris,
Please make sure you are using the latest update to the topotools plugin from my home page, since that contains some bug fixes that didn’t make it into the VMD 1.9 release.
I will take a look at your files tomorrow.
One question: how do you know that the unexpected bonds do not exist in the data file?
Cheers,
Axel.
Axel, thanks for the tip. I’ll get a chance to update topotools tomorrow and have a look. As for the possibility that the data file actually contains the strange bonds…The test.data file doesn’t show that any of the 8 bond types listed include hydrogen-hydrogen bonds (the Bond Coeffs section lists the atom types included in each bond as a comment). Also, the Bonds section lists all bonds, including the atom type of the bond members. I know, there’s no guarantee that some indices didn’t get crossed up, but the data file would have to be pretty cracked in that case.
Chris
Axel, thanks for the tip. I'll get a chance to update topotools tomorrow and
have a look. As for the possibility that the data file actually contains the
strange bonds....The test.data file doesn't show that any of the 8 bond
types listed include hydrogen-hydrogen bonds (the Bond Coeffs section lists
the atom types included in each bond as a comment). Also, the Bonds section
lists all bonds, including the atom type of the bond members. I know,
there's no guarantee that some indices didn't get crossed up, but the data
file would have to be pretty cracked in that case.
i just checked the data file on my desktop and it appears to be correct
and doesn't show any of the funky bonds that you were seeing.
now, i have two questions about your input file:
1) you are writing a trajectory for a subset of atoms (group diffusant)
did you create a data file for reading the topology of only those
atoms/molecules, too?
2) how did you load this trajectory into VMD? VMD should have refused
to load a trajectory with a different number of atoms than the existing
topology. the color scheme in your snapshot indicates that you may
have loaded it as a "new molecule". at that point, however, all bets are
off, since then VMD will try to guess atoms from the trajectory file.
...and will fail unless you take special precautions, since all it knows
are the atom type numbers, which will be set to something similar to
carbon and _that_ will then be used to construct bonds on atom radii
vs distance and could thus very well explain the unusual and unexpected bonds.
there are two possible strategies that you can follow.
a) load the data file and do a: topo guessatom lammps data
and then create a selection for each group that you define
in the lammps input to write a dump file off and the do for each.
$sel1 writepsf group1.psf
b) use dump_modify element to set and save element names in
you custom dump and then hope that VMD gets the heuristic
bond guessing right as soon as it knows the elements (it does
most of the time). for that you need to compile/install an update
VMD molfile plugin for lammps trajectories available (in source) from here:
http://sites.google.com/site/akohlmey/software/lammps-icms
hope that helps.
cheers,
axel.
Axel, OK got it, thanks. You were indeed correct in 2): I did load the trajectory as a new molecule, mistakenly presuming that topology info imported from the data file would inform the interpretation of any new lammps trajectory files. Now I am clear that only the current molecule ‘known’ about the topology info.
BTW, I am using your replicate topotools command on a trajectory. Can I get more than one frame into the new repeated mol?
Chris
Axel, OK got it, thanks. You were indeed correct in 2): I did load the
trajectory as a new molecule, mistakenly presuming that topology info
imported from the data file would inform the interpretation of any new
lammps trajectory files. Now I am clear that only the current molecule
'known' about the topology info.
naw, visualization programs, as a general rule, are very dump.
they don't reason, but only look at what is there. associative
thinking is a specialty of humans. 
BTW, I am using your replicate topotools command on a trajectory. Can I get
more than one frame into the new repeated mol?
not with me adding some extra code to the plugin.
can't you just use the periodic display feature of VMD?
axel.