I ran a lammps to do the md calculations. Calculated one picosecond duration after stabilising an electrolyte crystal, however I have some strange errors in my log file.
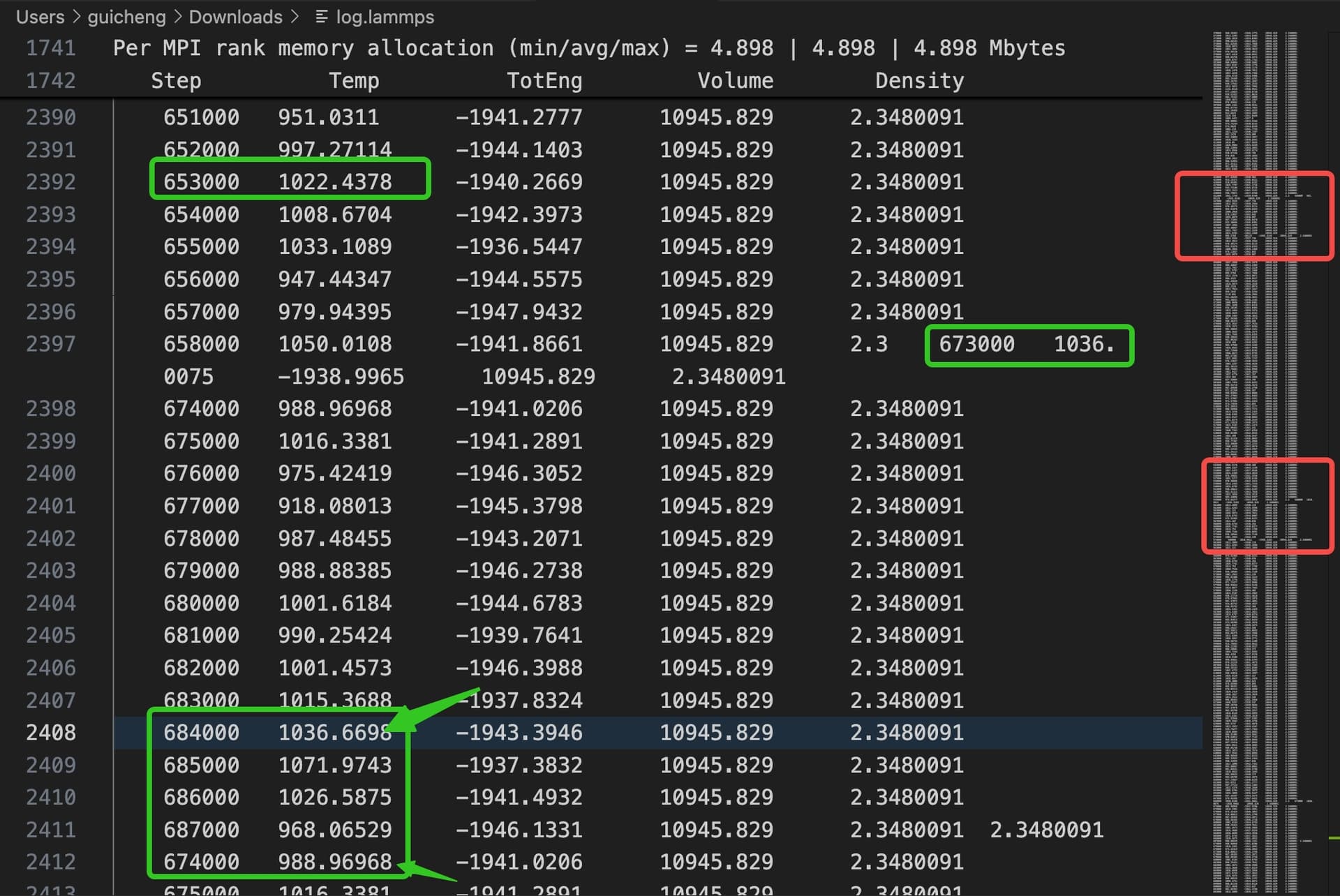
You can see data inserted haphazardly, disordered, misplaced, etc. in some data rows of the log file. I’m only running one MPI process, so theoretically there shouldn’t be multiple processes doing I/O at the same time to cause errors in the file. Please help!
ps. As I am a new user of the website, it prevent me from uploading files, so I’m sorry to use the coding block to show some of the code.They are the input file and submit file to the slurm system.If you need more information, plz let me know.
# LAMMPS input script for NVT run
# temperature
variable T equal 1000
# initialization
units metal
atom_style charge
neigh_modify delay 0 every 1
atom_modify map yes
newton on
# simulation box
box tilt large
read_data data_Li3YCl6-2_2_3
#group na type 1
# force field
pair_style mace no_domain_decomposition
pair_coeff * * /home/chenggui2/scratch/mace/model/MACE_MPtrj_2022.9.model-lammps.pt Li Y Cl
#----------------------Run Minimization-------------------------
reset_timestep 0
dump 1 all custom 100 ./dump.* id type x y z
thermo 1
thermo_style custom pe cella cellb cellc cellalpha cellbeta cellgamma vol density press etotal
fix 1 all box/relax aniso 0.0 vmax 0.001
min_style cg
minimize 1e-15 1e-15 5000 5000
# create v and ensemble setup
velocity all create $T 49459 mom yes rot yes dist gaussian
timestep 0.001 #set this to avoid atom loss in equilibration run
# equilibration run
reset_timestep 0
unfix 1
fix 2 all nvt temp 1 $T $(100.0*dt) # iso 1.0 1.0 $(1000.0*dt) #x 1 1 1 y 1 1 1 z 1 1 1
thermo 1000
run 10000
unfix 2
fix 3 all nvt temp $T $T $(100.0*dt) # iso 1.0 1.0 $(1000.0*dt)
thermo 1000
run 10000
# data gathering run
reset_timestep 0
timestep 0.001
## compute msd
#compute msdl na msd com yes
#variable msdx equal c_msdl[1]
#variable msdy equal c_msdl[2]
#variable msdz equal c_msdl[3]
#variable msdt equal c_msdl[4]
#
#fix 4 all vector 10 c_msdl[4]
thermo_style custom step temp etotal vol density #v_msdx v_msdy v_msdz v_msdt
thermo 1000
run 1000000
#!/bin/bash
#SBATCH --partition=gpu_v100s
#SBATCH -J md_nvt1000K
#SBATCH --nodes=1 # 1 computer nodes
#SBATCH --ntasks-per-node=4 # 1 MPI tasks on EACH NODE
##SBATCH --cpus-per-task=1 # 4 OpenMP threads on EACH MPI TASK
#SBATCH --gres=gpu:1 # Using 1 GPU card
#SBATCH --mem=20GB # Request 50GB memory
#SBATCH --time=3-00:00:00 # Time limit day-hrs:min:sec
#SBATCH --output=slurm.%j.out # Standard output
#SBATCH --error=err # Standard error log
module unload intelmpi
module load openmpi/4.1.5_gcc mkl/2020.4 cuda/12.1.0
### cudnn
export LD_LIBRARY_PATH="$LD_LIBRARY_PATH:/cm/shared/common_software_stack/packages/cudnn/cudnn-8.9.7.29_cuda12/lib"
export CPATH="$CPATH:/cm/shared/common_software_stack/packages/cudnn/cudnn-8.9.7.29_cuda12/include"
export LIBRARY_PATH="$LIBRARY_PATH:/cm/shared/common_software_stack/packages/cudnn/cudnn-8.9.7.29_cuda12"
source /home/chenggui2/scratch/anaconda3/bin/activate mace
export PATH=/home/chenggui2/scratch/lammps/build/:$PATH
mpirun -np 1 lmp -k on g 1 -sf kk -in in.nvt_1000K
LAMMPS (28 Mar 2023 - Development)
KOKKOS mode is enabled (src/KOKKOS/kokkos.cpp:107)
will use up to 1 GPU(s) per node
using 1 OpenMP thread(s) per MPI task
package kokkos
# LAMMPS input script for NVT run
# temperature
variable T equal 1000
# initialization
units metal
atom_style charge
neigh_modify delay 0 every 1
atom_modify map yes
newton on
# simulation box
box tilt large