Hello LAMMPS Community,
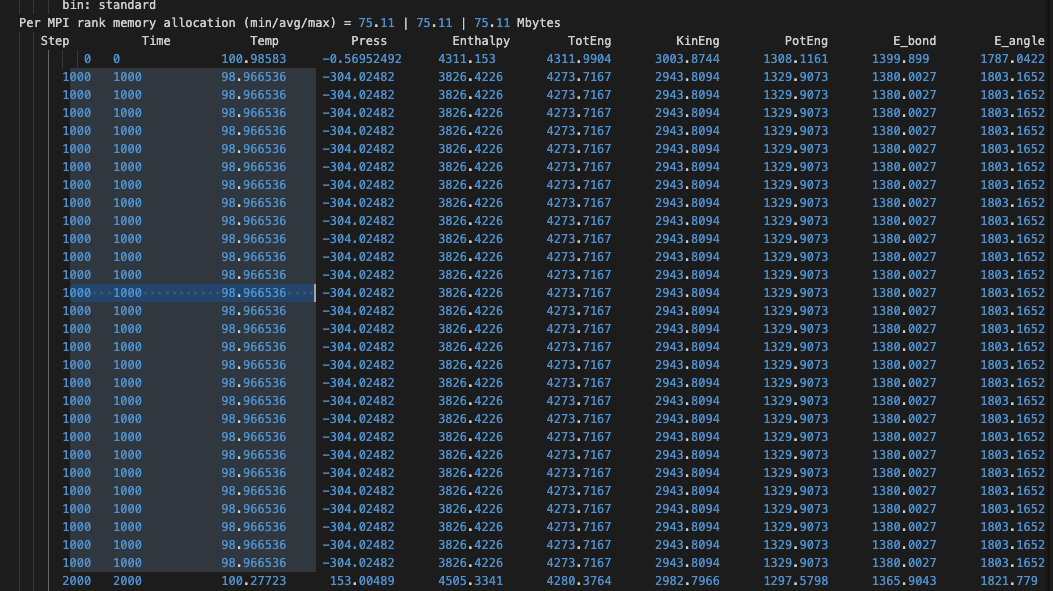
I’ve encountered an issue with my LAMMPS simulation and am seeking guidance on interpreting some output data in my log file. The snippet of the log file in question shows repeated entries, which I’m unsure how to analyze. Below is a sample of the output:
mailmaster://static.dashi.163.com/mailpart/1/133/D03065FD%40348B0740.3110016600000000.png
54000 54000 100.12154 -194.08587 3986.8042 4271.6327 2978.1657 1293.467 1372.6786 1819.3626 597.64531 10.621218 -3416.4145 7363.0039 -6453.4302 0 100626.76 46.512659 46.512659 46.512659 1.1529129 -790.70765 -50.756109 259.20616 -253.678 153.33876 -372.23349 0.19475422
55000 55000 99.673074 68.214321 4349.565 4249.4793 2964.8258 1284.6535 1365.1496 1804.6386 599.31798 10.521366 -3416.138 7373.806 -6452.6421 0 100605.26 46.509346 46.509346 46.509346 1.1531593 133.8344 -266.56736 337.37592 1.8199575 94.654642 -150.6625 0.20687099
This timestep is repeated multiple times with minor variations, and I’m particularly puzzled about how to interpret the columns related to energies and forces, as well as how these values could inform me about the dynamics and stability of my system.
Could anyone please explain:
- The significance of these repeated entries and if they indicate any issues with simulation stability or configuration? What is the cause of repeated or confused time steps?
I downloaded the LAMMPS (7 Feb 2024 - Development)code from GitHub and compiled it. My CMake command is:
cmake -C ../cmake/presets/most.cmake -D PKG_GPU=on -D GPU_API=cuda -D BUILD_SHARED_LIBS=yes -DGPU_ARCH=sm_75 -D LAMMPS_MACHINE=mpi -D BUILD_OMP=on -DCMAKE_LIBRARY_PATH=/usr/local/cuda/lib64/stubs -DCUDPP_OPT=ON -DCMAKE_POSITION_INDEPENDENT_CODE=ON ../cmake
I ran a LAMMPS command for equilibrium simulation, and the run command was:
mpirun -n 28 lmp_mpi -sf gpu -pk gpu 1 omp 1 -in eq4.in -sc none -log log.lammps.
My GPU model is GeForce RTX 2080 Ti Rev. A.
Thank your replying, I will give the requsted message.
- *the output of
lmp_mpi -h
Large-scale Atomic/Molecular Massively Parallel Simulator - 7 Feb 2024 - Development
Git info (develop / patch_7Feb2024_update1-147-g4d89741d8c)
Usage example: lmp_mpi -var t 300 -echo screen -in in.alloy
List of command line options supported by this LAMMPS executable:
-echo none/screen/log/both : echoing of input script (-e)
-help : print this help message (-h)
-in none/filename : read input from file or stdin (default) (-i)
-kokkos on/off ... : turn KOKKOS mode on or off (-k)
-log none/filename : where to send log output (-l)
-mdi '<mdi flags>' : pass flags to the MolSSI Driver Interface
-mpicolor color : which exe in a multi-exe mpirun cmd (-m)
-cite : select citation reminder style (-c)
-nocite : disable citation reminder (-nc)
-nonbuf : disable screen/logfile buffering (-nb)
-package style ... : invoke package command (-pk)
-partition size1 size2 ... : assign partition sizes (-p)
-plog basename : basename for partition logs (-pl)
-pscreen basename : basename for partition screens (-ps)
-restart2data rfile dfile ... : convert restart to data file (-r2data)
-restart2dump rfile dgroup dstyle dfile ...
: convert restart to dump file (-r2dump)
-reorder topology-specs : processor reordering (-r)
-screen none/filename : where to send screen output (-sc)
-skiprun : skip loops in run and minimize (-sr)
-suffix gpu/intel/kk/opt/omp: style suffix to apply (-sf)
-var varname value : set index style variable (-v)
OS: Linux "Ubuntu 22.04.2 LTS" 5.15.0-91-generic x86_64
Compiler: GNU C++ 11.4.0 with OpenMP 4.5
C++ standard: C++11
MPI v1.0: LAMMPS MPI STUBS for LAMMPS version 7 Feb 2024
Accelerator configuration:
GPU package API: CUDA
GPU package precision: mixed
OPENMP package API: OpenMP
OPENMP package precision: double
OpenMP standard: OpenMP 4.5
Compatible GPU present: yes
FFT information:
FFT precision = double
FFT engine = mpiFFT
FFT library = FFTW3
Active compile time flags:
-DLAMMPS_GZIP
-DLAMMPS_SMALLBIG
sizeof(smallint): 32-bit
sizeof(imageint): 32-bit
sizeof(tagint): 32-bit
sizeof(bigint): 64-bit
Extension: .zst Command: zstd
Extension: .xz Command: xz
Extension: .lzma Command: xz
Extension: .lz4 Command: lz4
Installed packages:
AMOEBA ASPHERE BOCS BODY BPM BROWNIAN CG-DNA CG-SPICA CLASS2 COLLOID COLVARS
CORESHELL DIELECTRIC DIFFRACTION DIPOLE DPD-BASIC DPD-MESO DPD-REACT
DPD-SMOOTH DRUDE EFF ELECTRODE EXTRA-COMPUTE EXTRA-DUMP EXTRA-FIX
EXTRA-MOLECULE EXTRA-PAIR FEP GPU GRANULAR INTERLAYER KSPACE LEPTON MACHDYN
MANYBODY MC MEAM MESONT MISC ML-IAP ML-POD ML-SNAP MOFFF MOLECULE OPENMP OPT
ORIENT PERI PHONON PLUGIN POEMS QEQ REACTION REAXFF REPLICA RIGID SHOCK SPH
SPIN SRD TALLY UEF VORONOI YAFF
List of individual style options included in this LAMMPS executable
* Atom styles:
amoeba angle atomic body bond
bpm/sphere charge dielectric dipole dpd
edpd electron ellipsoid full hybrid
line mdpd molecular oxdna peri
smd sph sphere spin tdpd
template tri
* Integrate styles:
respa respa/omp verlet verlet/split
* Minimize styles:
:
- the input file and log file(Since the log file is very large, I deleted some of it):
eq4.in (1.6 KB)
eq4.log (1.2 MB)
Here is the explanation for the repeated lines.
When running the CMake configuration, cmake could not find a usable MPI library (possibly because you have only the MPI runtime and not the MPI development package installed) and thus has created a serial executable. You are not running in parallel, but 28 times the same simulation. His is why the output is repeated.
This is confirmed by the output in your log file:
Reading data file ...
orthogonal box = (-23.271464 -23.271464 -23.271464) to (23.271464 23.271464 23.271464)
1 by 1 by 1 MPI processor grid
reading atoms ...
10000 atoms
The numbers for the “MPI processor grid” when multiplied should result in the total number of processors used but in your case it is 1 and not 28.
To address this issue, you need to make a suitable MPI package available. You can test for it by enforcing and MPI parallel build during CMake configuration with -D BUILD_MPI=yes. CMake will fail unless a suitable MPI installation is found, the details of that are not a LAMMPS but a CMake issue and an issue of your machines configuration and installation.
Thank you very much, I will try to recompile.
Mind you it is not advisable to oversubscribe a single GPU 28 times like you are doing here.
Something like 4-8 should be sufficient and you will be hard pressed to get significant speedup after that. Specifically for a system with only 10000 atoms. If you have too few atoms per GPU kernel invocation (i.e. when running with 28 MPI processes), the GPU kernels will run inefficiently. Yet, the total overhead of launching the kernels will increase.
Thank you very much for your advice. I will reduce the process of MPI.