Dear LAMMPS users,
I am trying to simulate thermal flux through SiO2 crystal using AtC package. Below are the details of the problem I am facing:
Version of LAMMPS used : 1Feb14
Question regarding package : Atc
Type of phenomenon being simulated : Thermal flux in SiO2
Short description of problem : Simulation is not completed due to unknown error
Detailed description of problem:
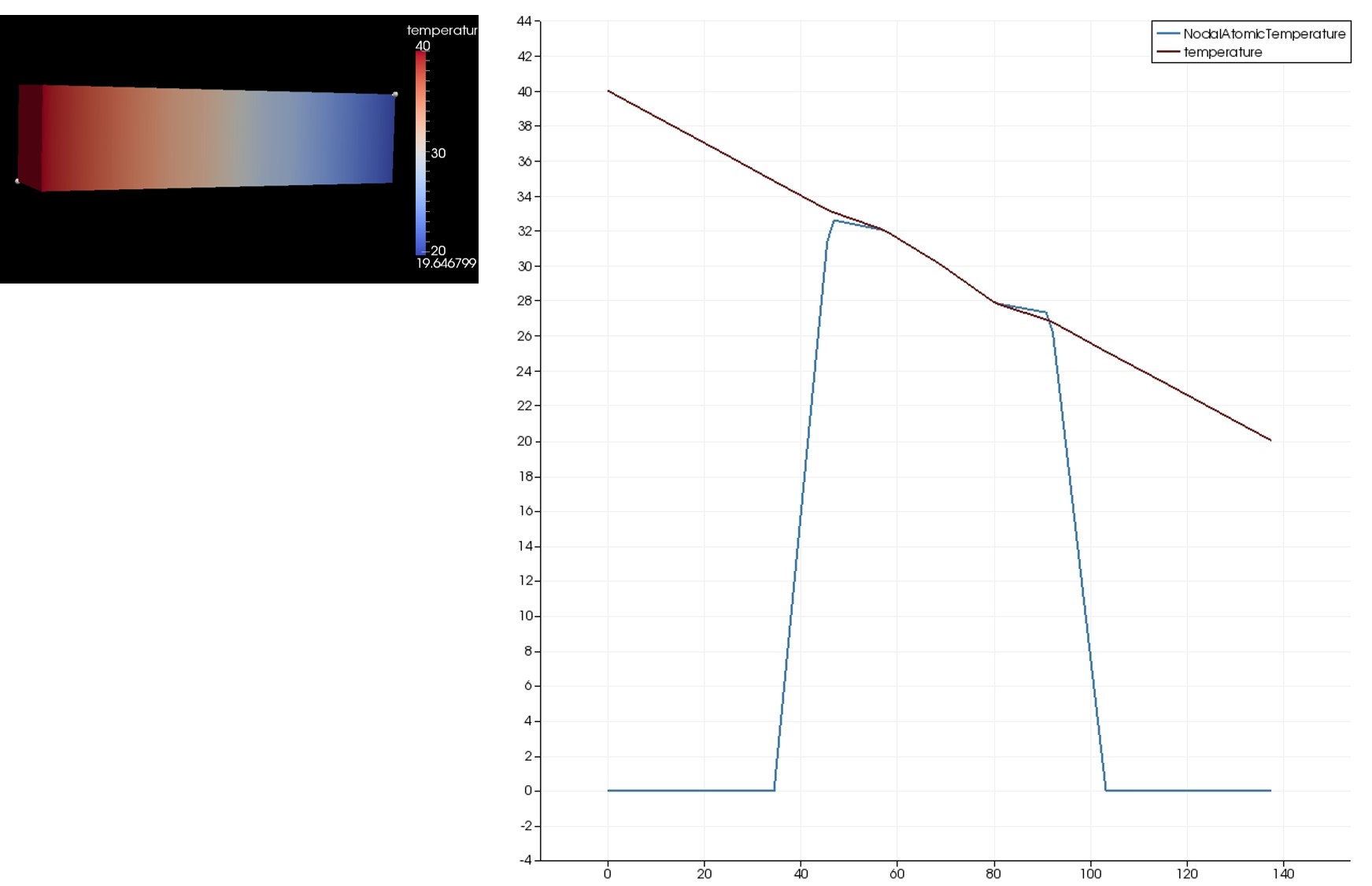
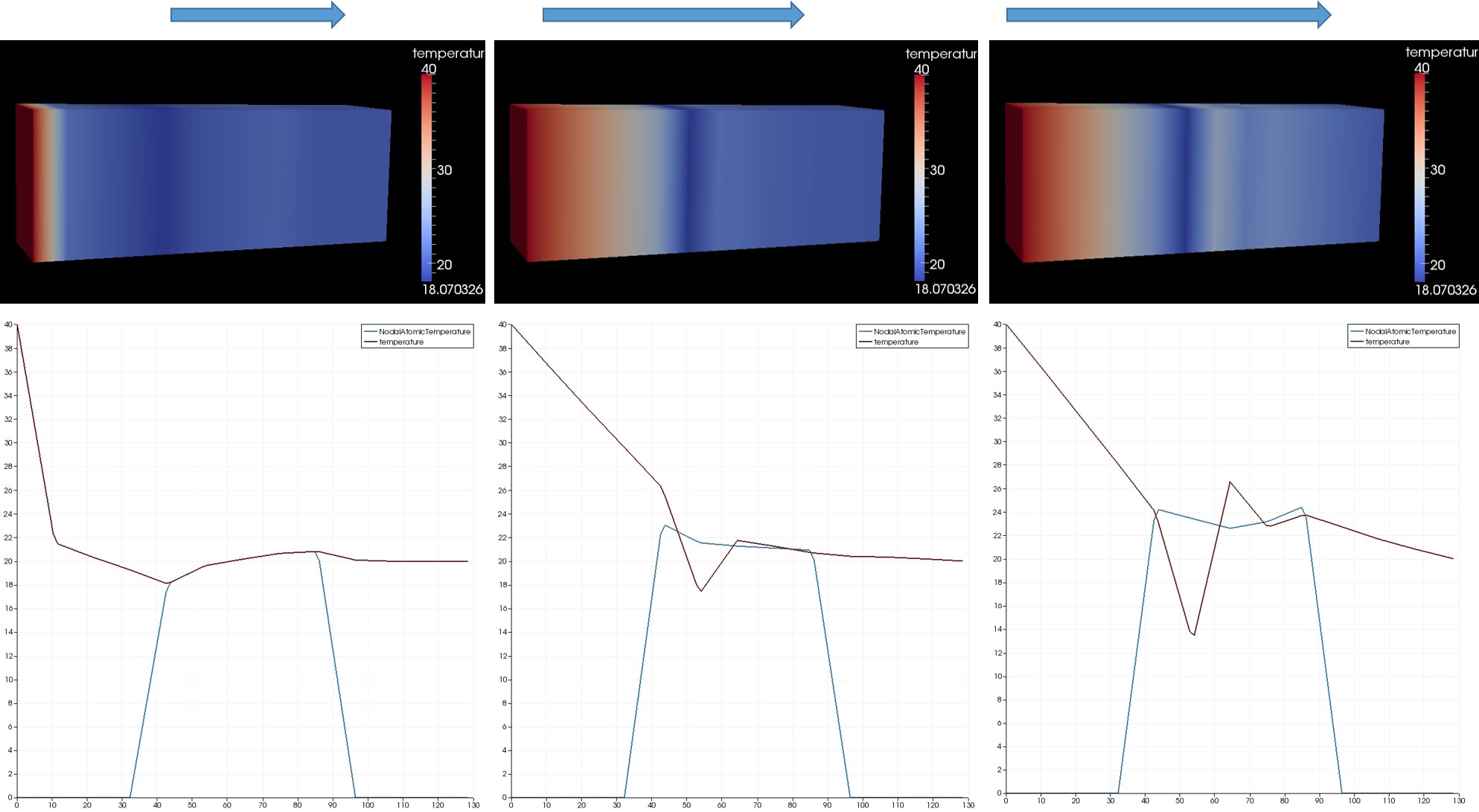
In order to simulate the thermal flux in SiO2 (alpha quartz), I modified the input file “in.bar1d_flux” in the example folder “thermo” under the “atc” examples.
Below is my modified input file where I am defining the SiO2 unit cell using the “lattice” command. My simulation gets terminated after 30 timesteps.
When I ran the simulation using the “-echo screen” option, the following line gets printed in the log file:
“DiagonalMatrix::inv(): (0,0)=0”.
I am not sure what this means.
Questions:
- Could you please suggest a way to specify the Silica unit cell that is acceptable for the AtC package?
- Why do I get the following warning - “ATC: WARNING: Cannot get number of atoms per cell from lattice” ?
- Is the warning the cause for the error or can I ignore it since its just a warning?
#Start of input script------
units metal
newton on
atom_style charge
dimension 3
boundary p p p
lattice custom 5.4054 origin 0.25 0.25 0.25 &
a1 0.90950000 0.00000000 .00000000 &
a2 0.00000000 0.78760000 .00000000 &
a3 .00000000 .00000000 1.00000000 &
basis 0.46990000 0.00000000 0.66666667 &
basis 0.00000000 0.46990000 0.33333333 &
basis 0.53010000 0.53010000 0.00000000 &
basis 0.41410000 0.26810000 0.78540000 &
basis 0.73190000 0.14600000 0.45206667 &
basis 0.85390000 0.58589999 0.11873333 &
basis 0.26810000 0.41410000 0.21460000 &
basis 0.58589999 0.85399999 0.88127777 &
basis 0.14600000 0.73190000 0.54793333
#region box prism 0 10 0 10 0 10 -0.4547 0 0
region simRegion block -12 12 -3 3 -3 3
region mdRegion block -5 5 -3 3 -3 3
create_box 2 mdRegion
create_atoms 1 region mdRegion &
basis 1 1 &
basis 2 1 &
basis 3 1 &
basis 4 2 &
basis 5 2 &
basis 6 2 &
basis 7 2 &
basis 8 2 &
basis 9 2
mass 1 28.09
mass 2 16.0
region mdInternal block -4 4 -3 3 -3 3
group internal region mdInternal
group ghost subtract all internal
pair_style lj/cut 2.5
pair_coeff * * 1.0 1.0 2.5
pair_coeff 1 2 1.0 2.0 3.5
pair_coeff 2 2 1.0 3.0 4.0
neighbor 5. bin
neigh_modify every 10 delay 0 check no
minimize 1.0e-4 1.0e-6 100 1000
#dump D1 all atom 100 dump.minbar1d
fix AtC internal atc thermal SiO2_thermal.mat
fix_modify AtC boundary ghost
fix_modify AtC time_integration fractional_step
ID part keywords nx ny nz region
fix_modify AtC mesh create 12 6 6 simRegion p p p
fix_modify AtC mesh create_faceset ibndy box -4.0 4.0 -INF INF -INF INF in
fix_modify AtC mesh create_faceset obndy box -4.0 4.0 -INF INF -INF INF outward
fix a temperature
fix_modify AtC fix temperature all 20.
turn on thermostat
fix_modify AtC control thermal rescale 10
dump D2 all atom 10 dump.bar1d
timestep 5
thermo 10
run 400
#end of input script------
Thank you,
Stephen Thomas.