Dear developer,
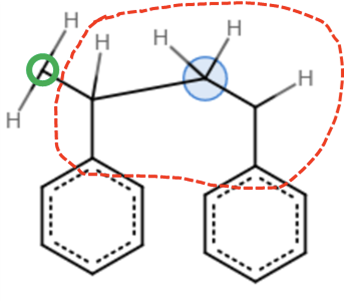
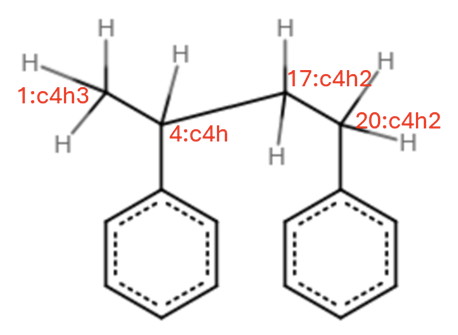
I’m trying to build a styrene dimer. In the esh file, If I give the full SMILES of the dimer, it works.
#!/usr/bin/env emc.pl
ITEM OPTIONS
replace true
density 0.1
number true
field opls-aa # type of FF to use
field_name emc-ff
field_location .
pdb_compress false
suffix "_linux_x86_64"
emc_execute true
emc_depth 6 # speed up ring searching
ITEM END
ITEM GROUPS
POL CC(c1ccccc1)CC(c1ccccc1)
ITEM END
ITEM CLUSTERS
pol POL,1
ITEM END
But If I treat the styrene dimer as a polymer and make use of the POLYMERS feature, emc failed with atom typing error
#!/usr/bin/env emc.pl
ITEM OPTIONS
replace true
density 0.1
number true
field opls-aa # type of FF to use
field_name emc-ff
field_location .
pdb_compress false
suffix "_linux_x86_64"
emc_execute true
emc_depth 6 # speed up ring searching
ITEM END
ITEM GROUPS
INI *[H]
STY *CC(*)c1ccccc1,1,INI:1,1,STY:2
TER *[H],1,STY:2
ITEM END
ITEM CLUSTERS
pol alternate,1
ITEM END
ITEM POLYMERS
pol
1 STY,2,INI,1,TER,1
ITEM END
EMC failed with the following error
Info: field = {mode -> apply, style -> none, error -> true, debug -> false,
check -> {atomistic -> true, charge -> true}}
Info: applying './emc-ff.prm'
Warning: no rule found for {group, site} = {STY, 0}.
Warning: no rule found for {group, site} = {STY, 3}.
Error: core/fields.c:433 FieldsApply:
Missing rules.
Program aborted.
In these two cases, the same top file is used for atom typing.
ITEM RULES
# id type element residue atom charge rule
1 c3 C - - 0 C(C(H)(H)(C))(^6c(:^6c)(:^6c))(H)(H)
2 c3 C - - 0 C(C(H)(H)(H))(^6c(:^6c)(:^6c))(H)(C(C)(H)(H))
3 c3 C - - 0 C(C(^6c)(H)(C))(H)(H)(H)
4 c3 C - - 0 C(C(^6c)(H)(H))(H)(H)(C(C)(^6c)(H))
5 ca C - - 0 ^6c(:^6c(:^6c)(H))(:^6c(:^6c)(H))(H)
6 ca C - - 0 ^6c(:^6c(:^6c)(H))(:^6c(C)(:^6c))(H)
7 ca C - - 0 ^6c(:^6c(C)(:^6c))(:^6c(:^6c)(H))(H)
8 ca C - - 0 ^6c(C(C)(H)(C))(:^6c(:^6c)(H))(:^6c(:^6c)(H))
9 ca C - - 0 ^6c(C(C)(H)(H))(:^6c(:^6c)(H))(:^6c(:^6c)(H))
10 ha H - - 0 H(^6c(:^6c)(:^6c))
11 hc H - - 0 H(C(C)(H)(C))
12 hc H - - 0 H(C(C)(H)(H))
13 hc H - - 0 H(C(C)(^6c)(C))
14 hc H - - 0 H(C(C)(^6c)(H))
ITEM END
Considering that these two esh files are describing exactly the same molecule, I cannot figure out why one works while the other does not.
I have attached the esh files and corresponding top and prm files for your reference.
emc-styrene.zip (2.4 KB)