Hello Dear Lammps Experts and Users,
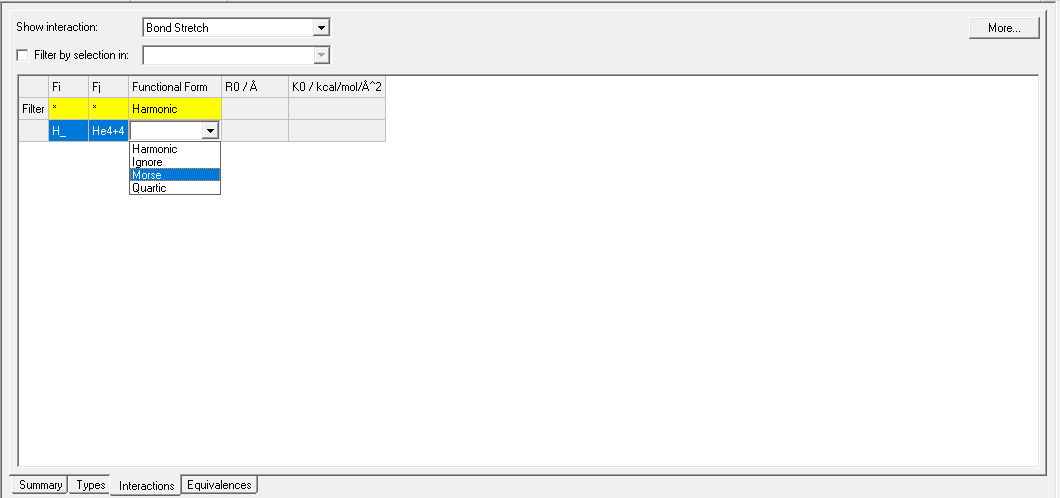
Nowadays, I try to implement UFF4MOF force field parameters into *.off file to usage in Forcite and Sorption Modules in Materials Studio (Figure-1 and Figure-2). However, true parametrization has to be done which is non-trivial task. Recently, I have found a way which has been used since many years. In this way, cif2lammps software converts the atomic position knowledge (from the cif files) into the force field terms like seen in Figure-3 (MOF-5 or IRMOF-1).
As you see from the Figure-3, there are two bond coeffs for the same C_R C_R interactions with different bond orders.
4 391.66951 1.45800 # C_R C_R bond order=1.0
6 462.65505 1.37926 # C_R C_R bond order=1.5
Probably, one of the C_R atoms is not true because the bond has different bond order.
This is important because I can not enter the same interaction to update *.off file. I have to make them different from each other.
In this direction, I have two questions.
-
Am I right to be in confusion whether they are the same or different C_R atoms?
-
If I am right, how to cope with it?
If you help me, I would appreciate.
Best regards
This is likely insufficient, since crystallographic .cif files usually do not contain bond information nor do they contain force field atom typing. So there is a lot of information missing for a classical force field calculation.
Why not? You can have different types of bonds between carbon atoms. Like an aliphatic C-C single bond or an aliphatic C=C double bond or an aromatic C-C bond in a phenyl ring. While those are all carbon atoms, they are usually given different atom types and partial charges in the force field, and the bonds also have different bond types (which are 4 and 6, respectively in your example).
You have to follow the exact definition of the force field you want to apply on how to assign force field settings. That is not something that is easily done from remote, so you better find yourself a tutor with experience in this to teach you how to do it and also teach you how force fields work.
You may want to check out the following: View article
Which contains an introduction for classical force fields written for beginners with a materials science background.
Thank you very much for your answer. I saw my mistake and corrected it.