Dear LAMMPS User:
I would like to make the random rough surface of fcc-Nickel.
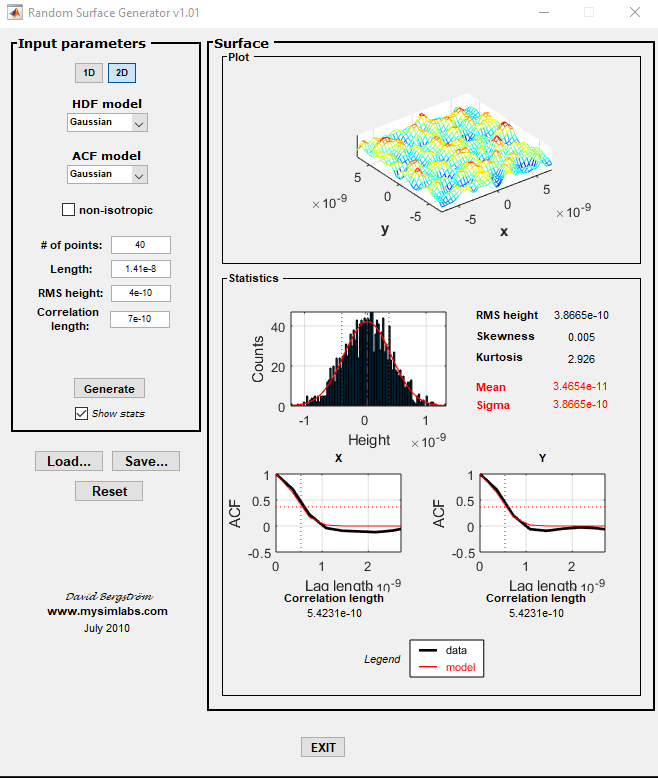
Firstly, I use the open accessible code to generate the rough surface. The details were in the snapshot. After saving the file, the positions for each point looks like square lattice arranged.
Secondly, I make a atomic block of fcc crystal structure (Nickel).
I would like to know whether it has any procedure to use the generated random surface to cut the block. Then, we can get the random rough surface of fcc-Nickel.
Thank you for any suggestions in advance.
Best wishes
Liyi

Dear LAMMPS User:
I would like to make the random rough surface of fcc-Nickel.
Firstly, I use the open accessible code to generate the rough surface. The details were in the snapshot. After saving the file, the positions for each point looks like square lattice arranged.
Secondly, I make a atomic block of fcc crystal structure (Nickel).
I would like to know whether it has any procedure to use the generated random surface to cut the block. Then, we can get the random rough surface of fcc-Nickel.
i assume "it" in this statement means LAMMPS. the general answer to
that is: anything that can be done with LAMMPS is documented in the
manual. we do not add features to LAMMPS without requiring it being
documented. so if you cannot find anything in the manual, it likely
means, that no such feature exists. sometimes, one has to "think
around the corner", and build a desired feature from combining
multiple features in LAMMPS.
Thank you for any suggestions in advance.
i don't think that this can be established without some programming. i
see different ways to implement this.
1) write a region style that can read and parse the generated rough
surface file and then select atoms that are either above or below the
(interpolated) surface in a given direction.
2) write a custom tool that generates the desired lattice (or reads a
dump file generated by LAMMPS) and also reads and parses the surface
file and then outputs a data file with the selected atoms.
i also would not discard alternative routes, that can be implemented
completely through LAMMPS input script. a simple procedure could be:
- create a "p p s" box of fcc-nickel atoms
- define a region of suitable at the top and use delete_atoms porosity
on that region to delete atoms at randomized positions
- run a minimization which should attract all atoms in the porous
region to the bulk. note the use of "s" boundary in this direction to
avoid interactions across pbc).
- equilibrate to the desired temperature.
you can control the roughness through the height of the region where
you delete atoms and the porosity parameter, and also through the
temperature at which you begin equilibration.
axel.
Dear Lammps Users and Prof. Axel.
Thank you very much for your reply.
You have mentioned three types of methods to make the simulation box with random roughness surface.
Usually, I create the simulation system by making a “DATA” file. I want to make the random rough surface starting with a smooth surface( I have already make the file with smooth Nickel surface). Could you please suggest which method will be more effective as you talked before? To be honest, I was trying to follow the procedures you points. However, i am still confused with some parts.
- write a region style that can read and parse the generated rough
surface file and then select atoms that are either above or below the
(interpolated) surface in a given direction.
Q: How is it possible to make region style than can read and generated rough if I don’t read the random surface through “Data file”?
- write a custom tool that generates the desired lattice (or reads a
dump file generated by LAMMPS) and also reads and parses the surface
file and then outputs a data file with the selected atoms.
Q: Yes, I can generate the desired lattice file (smooth Nickel surface).But, how to read and parses the surface
file and then outputs a data file with the selected atoms. How to define the function to follow the gaussion random distribution?( It might need random() function with formula. )
- completely through LAMMPS input script
For this method, do I need read smooth surface first?
Thank you very much for your precious time .
Best wishes
Liyi
Axel Kohlmeyer <[email protected]> 于2018年12月28日周五 上午2:57写道:
Dear Lammps Users and Prof. Axel.
Thank you very much for your reply.
You have mentioned three types of methods to make the simulation box with random roughness surface.
Usually, I create the simulation system by making a "DATA" file. I want to make the random rough surface starting with a smooth surface( I have already make the file with smooth Nickel surface). Could you please suggest which method will be more effective as you talked before? To be honest, I was trying to follow the procedures you points. However, i am still confused with some parts.
i have tried neither of those and which is the most effective route
depends on your specific needs, your general scientific skill level,
your programming skill level and your creativity and prudence. i don't
know you, so i cannot tell which to recommend.
1) write a region style that can read and parse the generated rough
surface file and then select atoms that are either above or below the
(interpolated) surface in a given direction.
Q: How is it possible to make region style than can read and generated rough if I don't read the random surface through "Data file"?
through c++ programming, of course. the basics on how to modify LAMMPS
are here: LAMMPS Molecular Dynamics Simulator
and the specific information pertinent to region styles here:
LAMMPS Molecular Dynamics Simulator
2) write a custom tool that generates the desired lattice (or reads a
dump file generated by LAMMPS) and also reads and parses the surface
file and then outputs a data file with the selected atoms.
Q: Yes, I can generate the desired lattice file (smooth Nickel surface).But, how to read and parses the surface
file and then outputs a data file with the selected atoms. How to define the function to follow the gaussion random distribution?( It might need random() function with formula. )
that depends on your (skript) programming skills and creativity. i can
imagine many different ways using different programming languages. the
general idea is the same as with the custom region style: you either
read or generate coordinates and determine whether they are on one or
the other side of the generated surface and then discard the ones you
don't want, and write the rest to a data file in the given format.
3) completely through LAMMPS input script
For this method, do I need read smooth surface first?
read or generate directly doesn't make a difference.
axel.
Dear Lammps User:
Thank you very much for your detailed explanation above.Now I focused on the third method which generate the random rough surface completely through LAMMPS input scripts.
You mentioned the steps like below.
- create a “p p s” box of fcc-nickel atoms
- define a region of suitable at the top and use delete_atoms porosity
on that region to delete atoms at randomized positions
- run a minimization which should attract all atoms in the porous
region to the bulk. note the use of “s” boundary in this direction to
avoid interactions across pbc).
- equilibrate to the desired temperature.
For the third step (in red color), as I understood from your state: I should fix the bottom surface and minimize the top part (after delete the atoms in the top region).However, according the minimize command manual, it can not work together with fix command.
So I switch to run nvt for the top atoms, while the bottom atoms are fixed (can not move).And I settle the Temp to 0 K. (Here I paste my input file) I would like to know whether I can use this method to minimize the system ?
Two more questions are:
- After relaxing the system, does the distance between the atoms in the top region can be around the lattice length?
- Does lammps delete_atoms command can delete atoms with a lattice cell instead of a single atom? Actually, I need the random surface still in fcc structure only with different heights .
Thank you very much for your precious time.
Best wishes
Liyi
input file :
variable T equal 0.0001 # Temperature unit=K
variable P equal 1.0 #Pressure unit=atm
units real
atom_style full
dimension 3
boundary p p s
set up box
lattice fcc 3.567
region box block 0 {xx} 0 {yy} 0 ${zz} units box #71.3471.3435.67
create_box 1 box
create_atoms 1 box
mass 1 12.010700 # carbon
velocity all create $T 72489 rot yes dist gaussian
pair_style lj/cut 12
pair_coeff 1 1 0.070810 3.214000
neighbor 2.0 bin
neigh_modify delay 0 every 20 check no
define a region of suitable at the top and use delete_atoms propsity on that region
region mydeformV block 0 {xx} 0 {yy} {lz} {zz} units box
region myfixV block 0 {xx} 0 {yy} 0 ${lz} units box
delete_atoms porosity mydeformV 0.5 482793 compress no
group top region mydeformV
group bottom region myfixV
fix 1 top nvt temp $T $T 100.0
fix 2 bottom move linear 0.0 0.0 0.0 units box
thermo 100
dump 1 all custom 1000 dump.tmp id x y z
run 1000000
Axel Kohlmeyer <[email protected]> 于2018年12月31日周一 下午12:51写道:
Dear Lammps User:
Thank you very much for your detailed explanation above.Now I focused on the third method which generate the random rough surface completely through LAMMPS input scripts.
You mentioned the steps like below.
- create a "p p s" box of fcc-nickel atoms
- define a region of suitable at the top and use delete_atoms porosity
on that region to delete atoms at randomized positions
- run a minimization which should attract all atoms in the porous
region to the bulk. note the use of "s" boundary in this direction to
avoid interactions across pbc).
- equilibrate to the desired temperature.
For the third step (in red color), as I understood from your state: I should fix the bottom surface and minimize the top part (after delete the atoms in the top region).However, according the minimize command manual, it can not work together with fix command.
that is not correct. not all fixes work with minimization, but fix
setforce 0 0 0 will work. you should apply this to only the lower part
of the crystal block to allow for some reconstruction and relaxation.
So I switch to run nvt for the top atoms, while the bottom atoms are fixed (can not move).And I settle the Temp to 0 K. (Here I paste my input file) I would like to know whether I can use this method to minimize the system ?
this is *not* what i suggested and a bad idea. what you are asking
reveals a serious lack of fundamental knowledge about MD and atomic
simulations. this is not the place to learn this and i don't have the
time to teach you.
Two more questions are:
1) After relaxing the system, does the distance between the atoms in the top region can be around the lattice length?
2) Does lammps delete_atoms command can delete atoms with a lattice cell instead of a single atom? Actually, I need the random surface still in fcc structure only with different heights .
it should be *very* obvious to you, that the second point is
conflicting with the aforementioned reconstruction and also with
equilibration/relaxation. i personally also consider this a not so
meaningful requirement (how do you get a rough surface? most certainly
not by methods that leave a perfect crystalline pattern behind), but
that is not my choice.
if you want to create such a geometry, you have to write your own tool
to build the structure. as mentioned before, this requires some
programming.
axel.