Dear Concerns
I am new in Lammps. Currently I am trying to fix room temperature (300K) of my substrate and apply velocity on particle. for this I tried several ways to fix my problem. I have two parts of simulation. (1) set room temperature (2) apply velocity in restart file. Here I am describing…
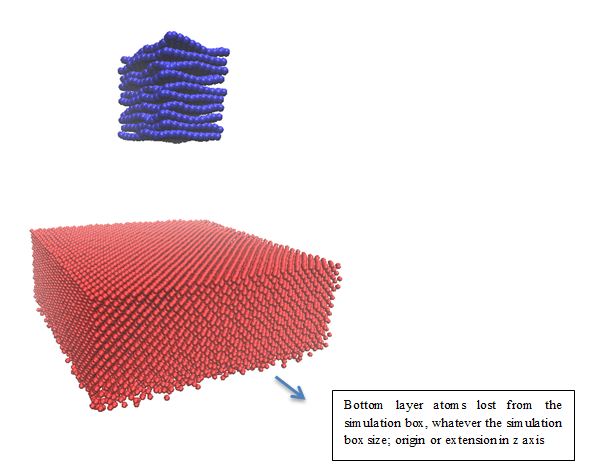
Case 1: I tried with NVT with gaussian distribution to fix room temperature. But found after some time steps Cu atoms are blowing from lower parts of simulation box, which I have attached in pic 1.
velocity all create 300.0 3213112 mom yes rot yes dist gaussian
fix 2 all nvt temp 300 300 100
Case 2: Then I tried with NVT and Langevin. I found same as case 1.
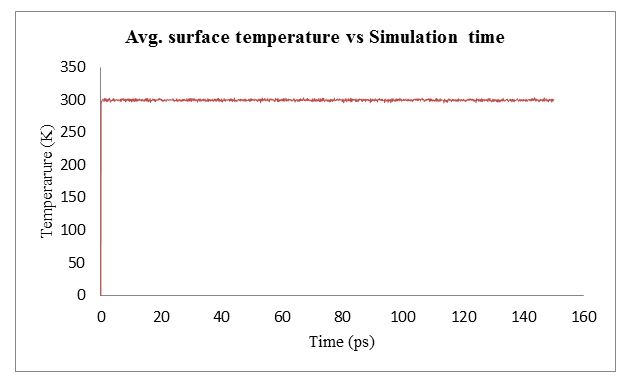
Case 3: Finally I tried with NVE and langevin. In this case I got good result and atoms are not blowing (see the attached pic 2 for temperature profile) below also input script
LAMMPS Input File
---------- Initialize Simulation ---------------------
units metal
dimension 3
boundary p p f
atom_style full
---------- Create Atomistic Structure ---------------------
read_data system1.LAMMPS05
group substrate type 1
group graphite type 2
region wall block INF INF INF INF -8.1500 -7.1529 units box
group bottom region wall
region middle block INF INF INF INF -7.1529 7.287090 units box
group stable region middle
region upper block INF INF INF INF 7.287090 106.802002 units box
group top region upper
region upper2 block 0.173601 99.448601 -0.966524 98.308502 28.947100 30.752100 units box
group surface region upper2
#fix 1 stable setforce 0.0 0.0 0.0
---------- Define Interatomic Potential ---------------------
pair_style hybrid eam lj/cut 8.0 airebo 4.0 0 0
pair_coeff * * airebo CH.airebo NULL C
pair_coeff 1 1 eam Cu_u3.eam
pair_coeff 1 2 lj/cut 0.00996 3.225
neighbor 2.0 bin
neigh_modify delay 10 check yes
---------- Run Minimization ---------------------
timestep 0.001
fix 2 substrate nve
#velocity all create 300.0 5812775 mom yes rot yes dist gaussian
#fix 2 all npt temp 300 300 100 iso 0 0 100
fix 3 substrate langevin 300 300 0.01 699483
#fix 3 all nvt temp 300 300 100
thermo 100
compute surface_temp substrate temp
compute surface_ke substrate ke
thermo_style custom step c_surface_temp c_surface_ke lx ly lz etotal
#thermo_style custom step temp ke pe lx ly lz etotal
#thermo_modify lost ignore
dump 1 all xyz 500 system_1313.xyz
restart 150000 deposite.restart
run 150000
Case 4: With this restart file I tried to apply velocity in z axis. But I observing some atoms lost from the simulation box like case 1. Here is my input file for apply velocity
LAMMPS Input File
---------- Initialize Simulation ---------------------
units metal
dimension 3
boundary p p f
atom_style full
---------- Create Atomistic Structure ---------------------
lattice fcc 1.0
read_restart deposite.restart.150000
fix 1 stable setforce 0.0 0.0 0.0
---------- Define Interatomic Potential ---------------------
pair_style hybrid eam lj/cut 8.0 airebo 4.0 0 0
pair_coeff * * airebo CH.airebo NULL C
pair_coeff 1 1 eam Cu_u3.eam
pair_coeff 1 2 lj/cut 0.07 3.225
neighbor 2.0 bin
neigh_modify delay 10 check yes
---------- Run Minimization ---------------------
timestep 0.001
#velocity top create 300.0 3213112 mom yes rot yes dist gaussian
#fix relax top nvt temp 300 300 0.01
fix 2 substrate nve
thermo 100
compute surface_temp surface temp
compute surface_ke surface ke
thermo_style custom step c_surface_temp c_surface_ke lx ly lz etotal
#thermo_style custom step temp ke pe lx ly lz etotal
velocity graphite set 0.0 0.0 -3.5
dump 1 all xyz 500 system_1_langen2.xyz
run 100000
For my simulation I don’t want to set z axis as periodic. At this moment I have no idea how I can apply velocity correctly. I am really expecting some fruitful comments or commands to solve my problem. Actually why atoms are blowing away ? Thanks in advance.
Regards
Nasim
RA at UOU