Dear Lammps-users,
I am trying to realize minimization using my own code in LAMMPS and I am using LAMMPS as an library.
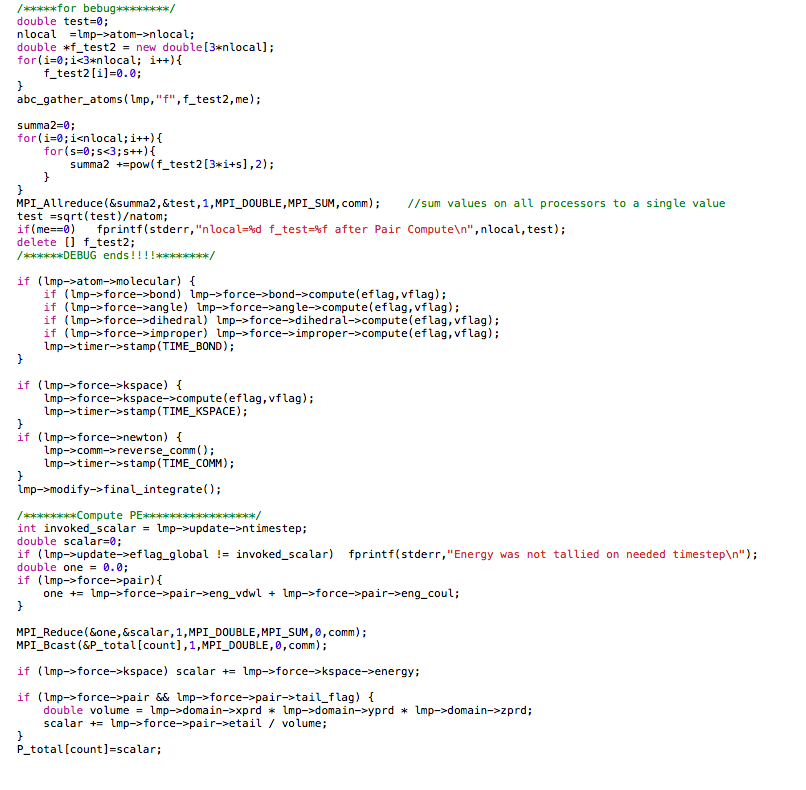
The code is similar to the min.cpp. In the minimization loop, I first evaluate the force on each atom and then calculate the new position for each atom. Atoms are moved to the new position. In order to evaluate energy and force of the system with new coordinates, I write a function similar to the energy_force() in min.cpp as can be seen in the attachment.
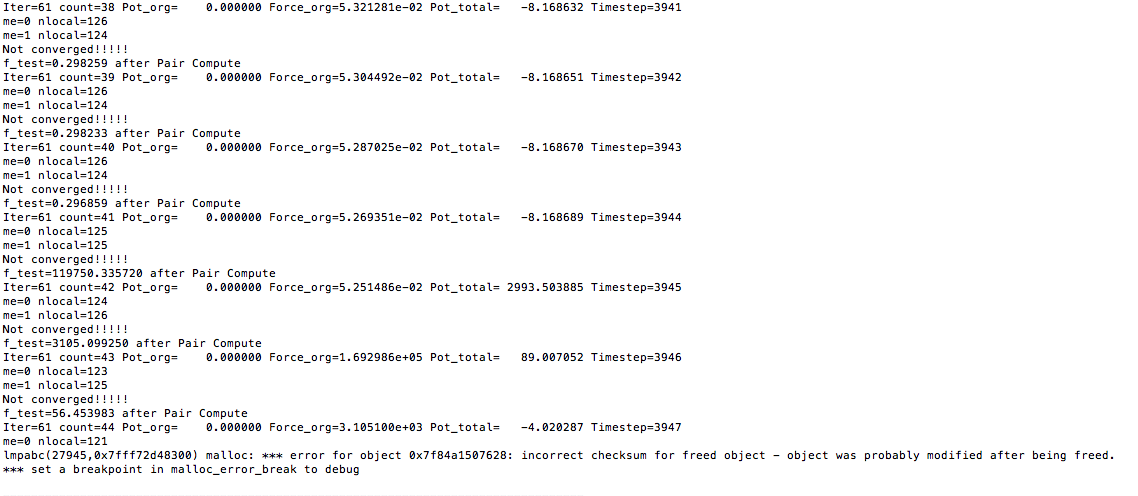
The code works well with one processor. However, when I run the code with two cores (parallel), the force and potential energy becomes wired when there is atom exchange between processors. The outputs are attached here.
I think the neighbor is not correctly built after atom exchange and as a result the force and energy evaluation is wrong. How can I modify my code to correct it? Do I need to add any fix to the system before I run my minimization code? Many thanks!!!

 Sincerely,
Sincerely,
Vivian