Hi, I seem to lose atoms in the minimization step despite using the fix wall command, the idea behind the input file is to put a negatively charged polymer over a positively charged polymer. Can someone see any fault?
01_06_2022_2.txt (2.9 KB)
I used even the fix modify energy option, yes, yet I am missing atoms
co-ion.tmp (81 Bytes)
counter-ion_negative.tmp (79 Bytes)
counter-ion_positive.tmp (80 Bytes)
pe_negative.tmp (1.1 KB)
pe_positive.tmp (1.0 KB)
The Template files for anyone who is interested in re-runs,
ERROR: Cannot open file 01_06_2022_40l_del1.polymer: No such file or directory (src/read_data.cpp:331)
Last command: read_data 01_06_2022_40l_del1.polymer add append
01_06_2022_40l_del1.polymer (232.9 KB)
Have you visualized your system to see what happens before the lost atom error happens?
That usually should give a significant hint of what is going wrong.
Yes Sir, I did, nothing seems to be amiss, I have done this in single input file for longer production runs I have not experienced any problems. only when I try to split the process workflow I am getting this error. The single input file I have attached here
Polymer_nagal_2_out.txt (7.1 KB)
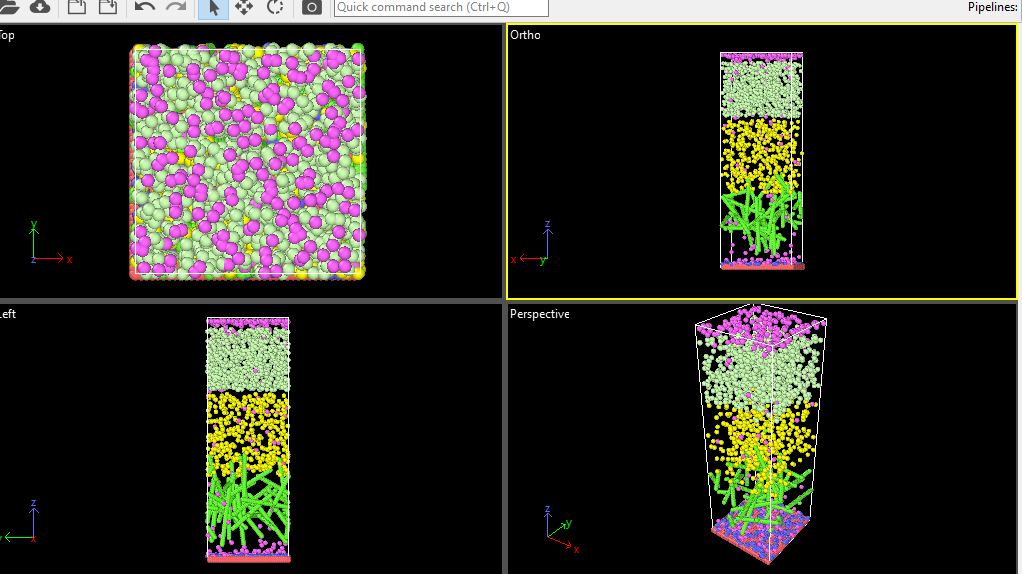
The above is the basic system. before minimization.
Look closer! What I see is that your bottom layer of atoms leaves the box after a few steps of minimization and that is because those are not immobilized.
Adding:
group wallzlo type 1
fix wallzlo wallzlo setforce 0.0 0.0 0.0
after the top wall fix addresses this issue.
Yes, Sir it worked, Thank you so much