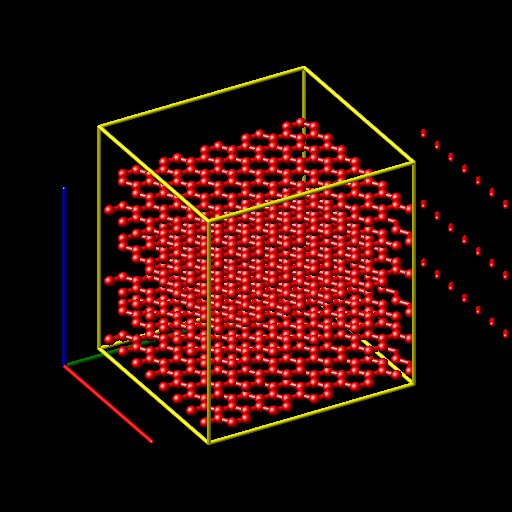
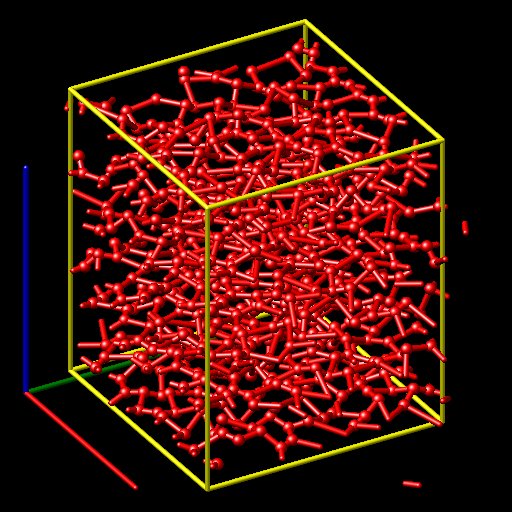
Recently I was trying to use LAMMPS to study graphite at high temperature(1800K) by using AIREBO potential. At the beginning, I set the temperature 300K. But the results showed that the structure was destroyed.(That could be imposible!). When I minimized the time step, the temperature seemed to be extremely high. I thought I made some mistakes in the input script. Maybe my graphite structure could be wrong. Could you please help me with this? Thanks!
Recently I was trying to use LAMMPS to study *graphite* at high
temperature(1800K) by *using AIREBO potential*. At the beginning, I set
the *temperature 300K*. But the results showed that *the structure was
destroyed.*(That could be imposible!). When I minimized the time step,
the temperature seemed to be extremely high. I thought I made some *mistakes
in the input script*. Maybe my graphite *structure could be wrong*. Could
you please help me with this? Thanks!
please look at the output that LAMMPS is producing?
you should see a warning about running a manybody potential with
bonds/angles/dihedrals defined.
that is the major hint to solving your problem: you are using a data file
with bonds and angles, but in AIREBO, those are all implicit through the
pair style. having bonds/angles in your topology will result in pairs of
atoms missing from the neighbor list and thus missing from the force
calculation.
the best approach is to remove those spurious bonds/angles from your data
file. a quick workaround would be to set all force constants for bonds and
angles to zero and use special_bonds lj/coul 1.0 1.0 1.0
Thanks for your kindly help! I removed bonds/angles from my data file. And it works very well. But there were still some problems.
When ‘velocity all create 3 482748 dist Gaussian’ in the input script, the graphite shake acutely. And the bonds became strange for the reason that some atoms moved to another side. Does that normal? When ‘velocity all create 3 482748 dist Gaussian’ were removed, the atoms just vibrated around its origin place.
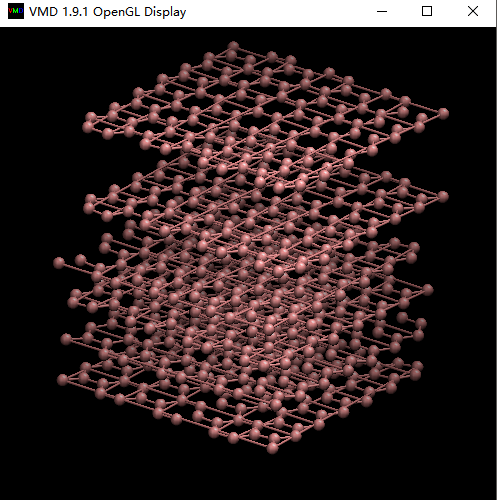
And the last picture was directly output from LAMMPS, there were no bonds between C atoms. But in VMD there were bonds between C atoms. Does that right?
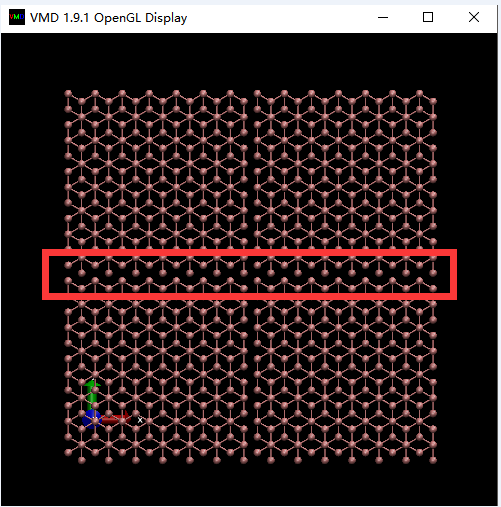
When I select periodic images to draw in the +x and +y direction, it seemed that there were no bonds connected between two C atoms. Does that normal?
Thanks for your kindly help! I removed bonds/angles from my data
file. And it works very well. But there were still some problems.
1) When 'velocity all create 3 482748 dist Gaussian' in the input
script, the graphite shake acutely. And the bonds became strange for the
reason that some atoms moved to another side. Does that normal? When 'velocity
all create 3 482748 dist Gaussian' were removed, the atoms just vibrated
around its origin place.
this seems primarily to be a visualization problem, but also note that
without the flag "zero linear" to the velocity command, you may be
introducing a net momentum to the system, which might cause it to start
drifting. please see the documentation of the velocity commands for details
and note the many discussions of drifting systems (and the flying icecube
syndrome) in the mailing list archives.
2) And the last picture was directly output from LAMMPS, there
were no bonds between C atoms. But in VMD there were bonds between C
atoms. Does that right?
yes, VMD uses some heuristic to guess bonding patterns, but in LAMMPS
there are definitely no explicit(!) bonds defined (as needed).
When I select periodic images to draw in the +x and +y
direction, it seemed that there were no bonds connected between two C
atoms. Does that normal?
yes, search the VMD mailing list archives. VMD has not been primarily
designed for condensed matter solids and its periodic display option works
by simple replicating the central cell. if you want display of bonds across
the boundaries in VMD, you have to replicate the coordinates and load such
an enlarged file.
In addition, you were using the REBO form by using “pair_style rebo” which only describes graphite well near equilibrium. For high T/P applications, you would want to use AIREBO.
Thanks for your detail reply! Your suggestions are super helpful! And I’ve got some reasonable results!
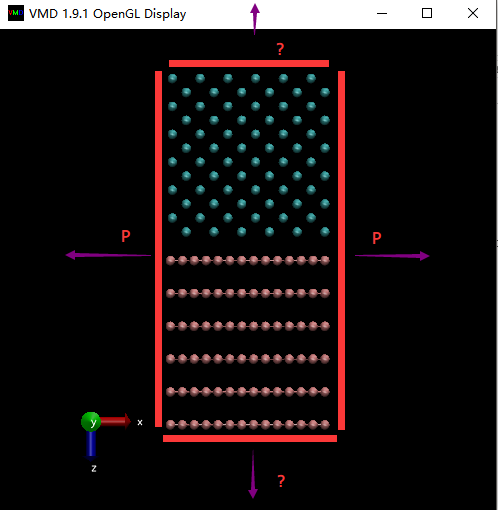
What I want to simulate is liquid iron diffusing and eroding the graphite at about 1800K. I really appreciate for that you have replied my question a month ago. And now I have some more detail questions to consult you.
I wonder if it is possible to make the graphite infinite in +z direction, and liquid iron infinite in –z direction. The other side can be set as period boundary condition.
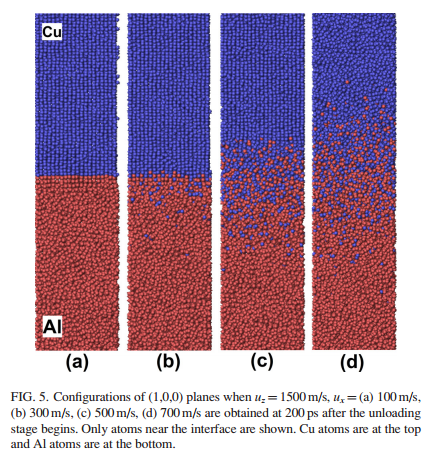
And I saw someone who simulated Cu-Al explosive welding process constraint the upper and lower several layers. And other sides were set as period boundary condition. Is it reasonable?
Best regards
Zhuang
School of Energy Science Engineering
Central South University, Changsha
Email:zhuangjiacai@…4215…
Thanks for your detail reply! Your suggestions are super helpful!
And I've got some reasonable results!
What I want to simulate is liquid iron diffusing and eroding the
graphite at about 1800K. I really appreciate for that you have replied my
question a month ago. And now I have some more detail questions to consult
you.
I wonder if it is possible to make the graphite infinite in +z
direction, and liquid iron infinite in –z direction.
no. that doesn't make sense anyway.
The other side can be set as period boundary condition.
And I saw someone who simulated Cu-Al explosive welding process
constraint the upper and lower several layers. And other sides were set as
period boundary condition. Is it reasonable?
it depends.
please note that none of this are really LAMMPS questions, but questions
about how to do your research. this is something that you first and
foremost should discuss with your adviser, infer from the published
literature and textbook material, just like everybody else.
Thanks for your reply. Do you mean that I cannot simulate the process which liquid iron diffusing in graphite? There are no one around me do the research about molecular simulation, and my adviser doesn’t familiar with that project.
I have read some articles about using Monte Carlo simulation to study dissolution of Graphite in Iron-Carbon Melts. V. SAHAJWALLA had done lots of these research. And here is one of her article: http://link.springer.com/article/10.1007/s11663-000-0036-9
When I see someone who use LAMMPS to simulate Cu-Al explosive welding process and get some results such as diffusion coefficients, I also want to use the same method to simulate liquid iron diffusing and eroding the graphite at about 1800K so as to differ from V. SAHAJWALLA’s work and verify her data.
Could you please explain the reason why I can’t do this with LAMMPS or MD for me? If it’s really too hard to solve this problem via LAMMPS or MD, I have to change my research.
Best regards
Zhuang
School of Energy Science Engineering
Central South University, Changsha
Email:zhuangjiacai@…4215…
Thanks for your reply. Do you mean that I cannot simulate the
process which liquid iron diffusing in graphite?
your question is ambiguous. that kind of simulation is certainly possible,
but whether you have the skills and ability to realize this is something i
cannot say.
There are no one around me do the research about molecular simulation, and
my adviser doesn't familiar with that project.
well, that is your problem, not mine. i don't consider it very smart to
start a project without having any practical knowledge about it and without
any form of competent supervision or training. it most certainly does not
entitle you to have somebody else compensate for such a misguided approach
to do research.
[...]
Could you please explain the reason why I can't do this with
LAMMPS or MD for me? If it's really too hard to solve this problem via
LAMMPS or MD, I have to change my research.
your problem is not a LAMMPS problem, but a problem in how you are
conducting research and how you are expecting to be able to just do
something that requires significant background knowledge and training. if
you do not have the necessary expertise at hand, you will either have to
find a collaborator that can provide the required supervision, advice and
training, or change your project to do something that you are properly
trained in.
Dear Axel
I really appreciate for your patient reply. Maybe I was wrong at the beginning. I will follow your suggestion and try my best to figure it out.
Thank you very much from the bottom of my heart!
Best regards
Zhuang
School of Energy Science Engineering
Central South University, Changsha
Email:zhuangjiacai@…4215…