Dear LAMMPS users,
I have been using nudged elastic band (NEB) method to find out minimum energy path (MEP) and energy barrier of transitions in HCP metals.
The NEB command in LAMMPS consists of N1 and N2 to represent initial NEB and barrier climbing NEB, respectively. I have noticed that after a certain number of iterations of initial NEB, the MEP converges and increasing the iterations doesn’t make any difference. However, when the barrier climbing NEB is increased, the energy barrier just keeps on increasing. The barrier climbing NEB is supposed to drive the replica with highest energy to the top of the saddle but it seems the highest energy replica just keeps getting more energy with the increment in iterations.
I have attached a screenshot with 4 MEPs, where the initial NEB is constant at 9000 timesteps but the barrier climbing NEB is increased from 2000 to 102000 timesteps and correspondingly, the peak energy is also increasing.
I don’t suppose there could be a problem with my script because the above mentioned observations seem to occur only while varying barrier climbing NEB, keeping everything else constant.
I couldn’t understand what is going on. Somebody please shed light on this.
I am using 12 December 2018 version of LAMMPS on my university computer.
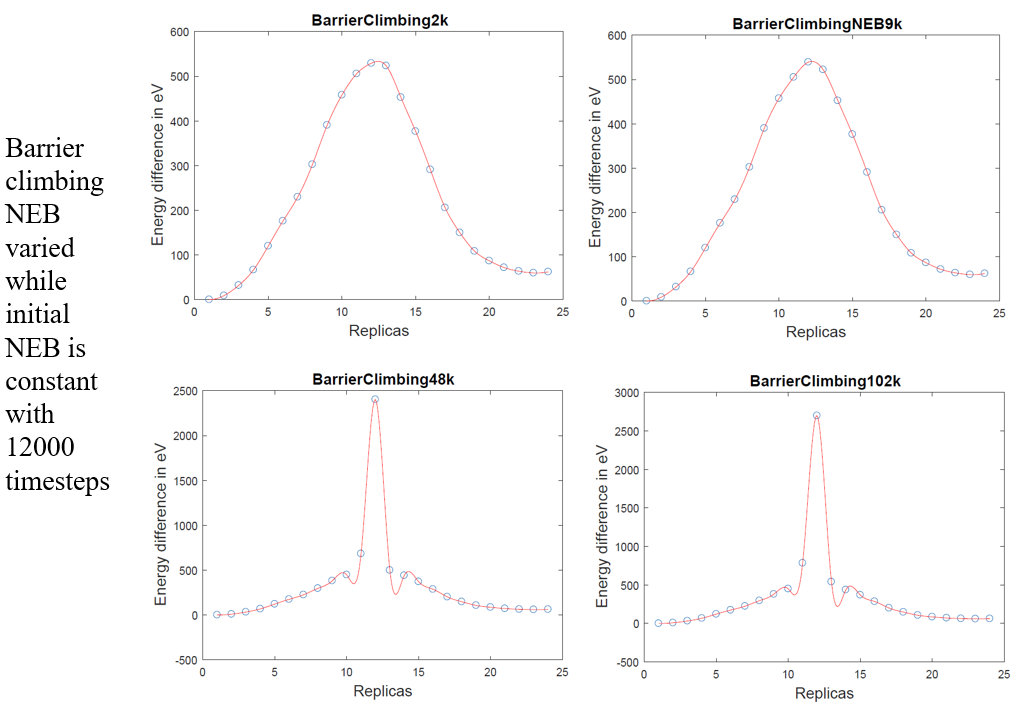
What is your expected activation energy for this process? How big is your system? Does the energy barrier ultimately converge? Try plotting the energy barrier as a function of minimization steps during the barrier climbing run to answer this question. 102000 min steps is not that many. My point is that this behavior may be perfectly fine and you should get more information. Your barrier climbing NEB is climbing the barrier, and appears to be converging, because when you go from 9000-48,000 steps, the barrier jumps by 2000 eV, as opposed to only going up by a couple hundred eV when going from 48,000-102,000 steps. The high energy region of your plot is very narrow, so one could even argue that this behavior is expected (the initial NEB could easily tunnel right through this region, not capturing it). Seems okay to me, but then again, I know nothing about your simulation.
-Keith
Hi Keith,
Thank you for your response.
Mine is a simulation box containing Ti bicrystal with around 48,000 atoms and they are undergoing twinning process. I think you are right in pointing out that the energy barrier might be converging as we increase the number of iterations. However, the energy barrier of around 2500 eV is extremely high for a twinning process.
Regards,
Deepesh
Hi Deepesh,
No problem!
What does 2500 eV mean in this case? Is it the absolute barrier in eV for your 48,000 atom system to undergo a twinning process? I am not familiar with this field of research, but energy is typically expressed relative to something. For a twinning process, should it be energy per unit area of twin surface formed? I have no idea. In any case, if the barrier really is too high, it could be due to a number of reasons, and sorry to say, but the onus is on you to investigate and narrow down the issue. It could be the interatomic potential, it could be how you set up your system, etc. It’s likely not a bug in the NEB implementation in LAMMPS. Once you’ve clarified the issue, you might have new questions that are in the scope of this mailing list, but as it stands, it’s not quite clear what your problem is, if you even have one, and how (or if) anyone here can help.
-Keith
Hi Keith,
I think I will need to check my simulation setup. Moreover, as you pointed out, it could be the interatomic potential as well. I will need to discuss with my advisor.
Thank you again for taking out time to provide me valuable feedback.
Regards,
Deepesh