Dear All,
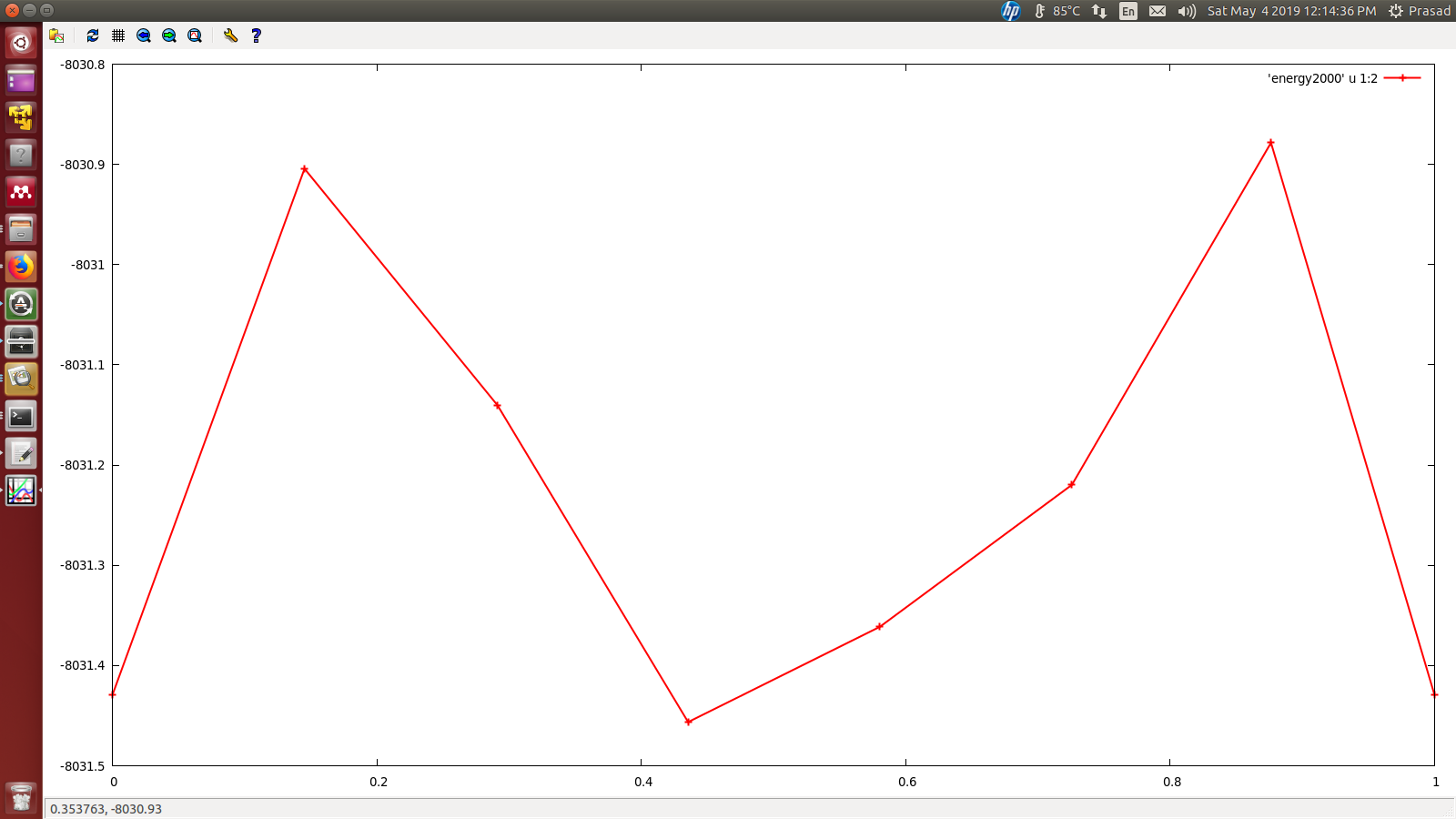
I am estimating activation barrier height for carbon atom diffusion from one octahedral site to other octahedral site for Fe-C system with NEB method in lammps. I am getting barrier height around 0.5 eV whereas, reported barrier height is around 0.85 eV.
Here is my script:
---------- Initialize Simulation ---------------------
clear
units metal
dimension 3
boundary p p p
atom_style atomic
atom_modify map array sort 0 0.0
variable u uloop 20
---------- Create Atomistic Structure ---------------------
lattice bcc 2.855
region box block 0.0 10.0 0 10.0 0 10.0
#create_box 2 box
Create Fe atoms
#create_atoms 1 box
Insert Carbon interstitial
#create_atoms 2 single 5.0 4.5 4.5
read_data initial.carbon
---------- Define Interatomic Potential ---------------------
pair_style eam/fs
pair_coeff * * Fe-C_Hepburn_Ackland.eam.fs Fe C
group carbon type 2
group iron type 1
#write_data initial.carbon
---------- Define Settings ---------------------
compute csym all centro/atom bcc
compute eng all pe/atom
compute eatoms all reduce sum c_eng
compute int_en carbon group/group iron
region surround block 3.5 6.5 3.5 6.5 3.5 6.5
group nebatoms region surround
group nonneb subtract all nebatoms
---------- Run Minimization before displacement---------------------
#minimize 1e-15 1e-15 5000 5000
reset_timestep 0
thermo 100
#thermo_style custom step pe lx ly lz press pxx pyy pzz c_eatoms
timestep 0.001
fix 1 all nve
fix 2 nonneb setforce 0.0 0.0 0.0
fix 3 nebatoms neb 10.0 parallel neigh perp 1.0
dump 1 nebatoms atom 10 dump.neb.$u
dump 2 nonneb atom 10 dump.nonneb.$u
min_style quickmin
neb 1e-15 1e-15 10000 10000 1000 final final.carbon
Please suggest, where am I going wrong.
I am attaching the curve for activation barrier height.