Dear lammps users
I used reax/c/bonds and reax/c/species in my input, and then the outcome was written into two different files. I have some basic questions about the commands and information in output files. I used following commands.
fix 3 all reax/c/species 10 10 100 species.tatb
fix 4 all reax/c/bonds 1000 bonds.reaxc
My questions are:
(1) According to the manual, the first command means that the bonder order value on timpstep within first 10,20,30….,80,90,100 will be used to compute the average bond-order for the species analysis output on timestep 100. However, I am confused about how to calculate the average bond order for next 100 timestep. One understanding is that the bonder order value on timpstep 110,120,130….,180,190,200 within next 100 tiemstep will be used to compute the average bond-order for the species analysis output on timestep 200. Or the bonder order value on timpstep 10,20,30….,80,90,100, 110,120,130,…180,190,200 will be used to compute the average bond-order for the species analysis output on timestep 200. Which one is right?
(2) If the first understanding in (1) is right, the second command means that only the bond order information on 1000th timestep is written into file. Is that right?
(3)Is there any criteria for choosing the parameter (I mean 10 10 100) for reax/c/species?
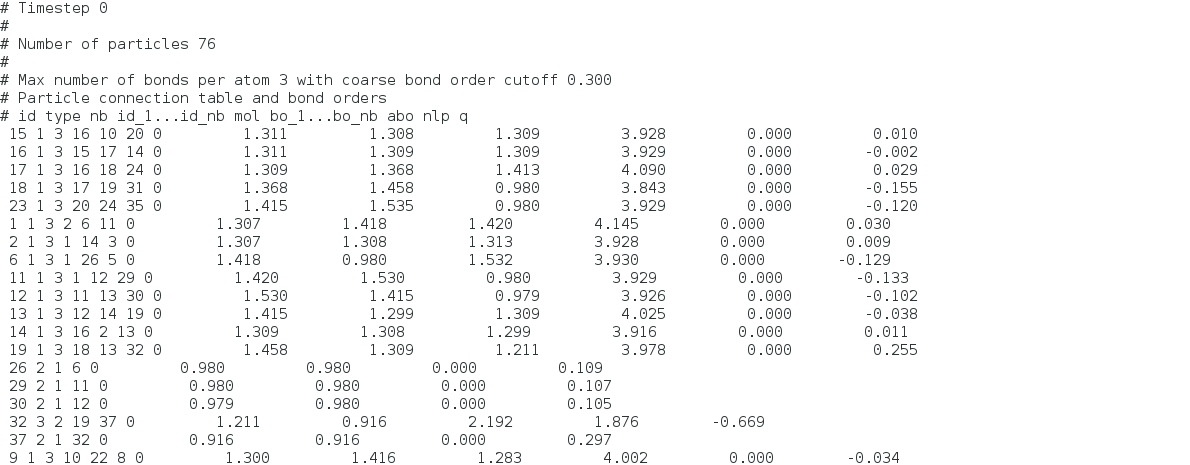
The first picture is the output reax/c/bonds. I understand some of items. For example, items give information about id of the atom and atoms that are bond with them from the first (id) to fifth item (id_nb). And bo_nb is bond order information. My question is
(4)What are “mol”, “abo” and nlp?
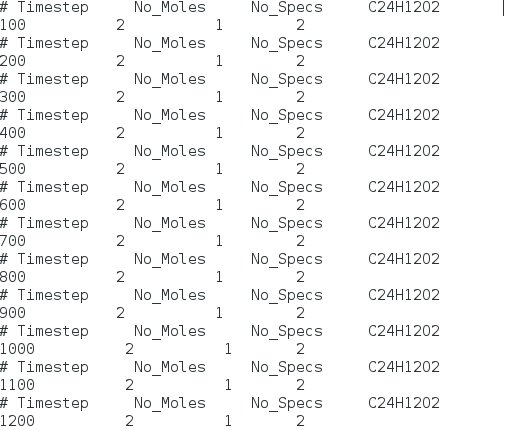
The second picture is output of reax/c/species. I know that “No_Moles” and “No_Spacs” are the number of molecules and species My question are
(5)Is there any difference between molecules and species?
(6)What “1” represents?
The third and fourth picture are my data file and input file. I am very sorry to ask such basic questions.
input.docx (13.8 KB)
corodata.pdf (35.6 KB)