Dear all,
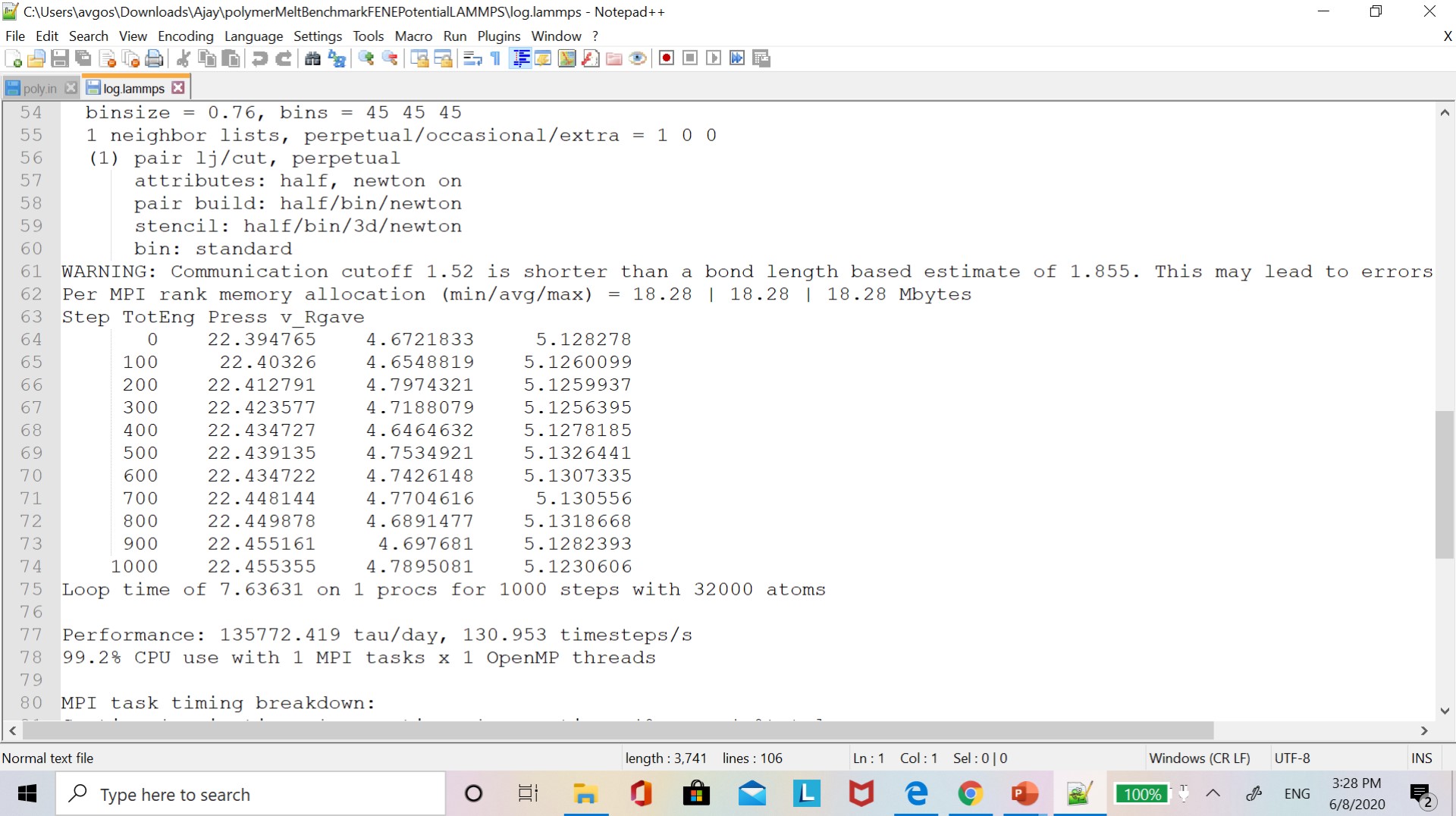
Currently, I was running a polymer melt benchmark study problem for 32000 atoms to calculate the average radius of gyration. Input files, log out files, and data files are given in the LAMMPS benchmark study folder. I am getting the same output log files after running simulation for 1000 steps. and I also calculated the average radius of gyration. But my radius of gyration value did not match with the radius of gyration values given in the LAMMPS manual (section 17.12.3).
Can you look into it? Why I am getting the wrong radius of gyration values?
For your reference, I am attaching an input script, a screenshot of my radius of gyration values, and a radius of gyration values given in the LAMMPS manual.
I am using the 15Apr2020 version of LAMMPS.
here the details of the simulation.
Bead-spring polymer melt with 100-mer chains and FENE bonds:
- 32,000 atoms for 100 timesteps
- reduced density 0.8442 (liquid)
- force cutoff of 2^(1/6) sigma
- neighbor skin = 0.4 sigma
- neighbors/atom = 5 (within force cutoff)
- NVE time integration
Due to limitation of size I am unable to attach the data file. but data file,i nput file is given in the LAMMPS benchmarks study folder (chain.data).
Thank,
Ajay
poly.in (664 Bytes)

log.lammps (3.65 KB)
Dear all,
Currently, I was running a polymer melt benchmark study problem for 32000 atoms to calculate the average radius of gyration. Input files, log out files, and data files are given in the LAMMPS benchmark study folder. I am getting the same output log files after running simulation for 1000 steps. and I also calculated the average radius of gyration. But my radius of gyration value did not match with the radius of gyration values given in the LAMMPS manual (section 17.12.3).
that is not an example for computing the radius of gyration, but an example for using compute chunk/spread/atom to add a force to molecules so that the radius of gyration will systematically shrink. your input only has the computation of the radius of gyration, but to see it shrink and thus get the same values as in the manual, you would also need to have the fix addforce line and all the computes it depends on.
Can you look into it? Why I am getting the wrong radius of gyration values?
because you didn’t pay attention to what it is that the example is explaining.
For your reference, I am attaching an input script, a screenshot of my radius of gyration values, and a radius of gyration values given in the LAMMPS manual.
I am using the 15Apr2020 version of LAMMPS.
here the details of the simulation.
Bead-spring polymer melt with 100-mer chains and FENE bonds:
- 32,000 atoms for 100 timesteps
- reduced density 0.8442 (liquid)
- force cutoff of 2^(1/6) sigma
- neighbor skin = 0.4 sigma
- neighbors/atom = 5 (within force cutoff)
- NVE time integration
Due to limitation of size I am unable to attach the data file. but data file,i nput file is given in the LAMMPS benchmarks study folder (chain.data).
this would be a very wasteful thing, since everybody that will be able to look into this and help you will have a LAMMPS installation and thus also the data file. please keep in mind that by sending an e-mail to the mailing list you are effectively sending out about a thousand e-mails and thus those e-mails to the list must not exceed certain limits. and it is pretty pointless to send people files they already have.
axel.