Dear all,
I am trying to investigate the interface between Fe and cementite. Although I faced no error, I am wondering if what I am doing is correct, in particular, regarding the equilibration part.
Here is the input I used.
dimension 3
units metal
atom_style atomic
boundary p p p
# Definition of useful variables
variable sqrt2 equal sqrt(2)
variable sqrt3 equal sqrt(3)
variable sqrt18 equal sqrt(18)
variable sqrt6 equal sqrt(6)
# Fe lattice param
variable aFe equal 2.8553
# cementite lattice params
variable acem equal 4.488
variable bcem equal 5.032
variable ccem equal 6.721
# Number of Fe cells in all directions (the reference)
variable ncellxFe equal 13
variable ncellyFe equal 30
variable ncellzFe equal 4
variable ncellxcem equal 11
variable ncellycem equal 29
variable ncellzcem equal 4
lattice bcc 2.8553 orient x 1 -1 0 orient y 1 1 1 orient z -1 -1 2 origin 0.0 0.0 0.0 spacing ${sqrt2} ${sqrt3} ${sqrt6}
region simdom block 0 ${ncellxFe} 0 ${ncellyFe} 0 12 units lattice
create_box 2 simdom
mass 1 55.847
mass 2 12.0111
pair_style meam
pair_coeff * * Fe3C_library_Liyanage_2014.meam Fe C Fe3C_Liyanage_2014.meam Fe C
# Cementite layer
# First we define the primitive cell of the cementite
# It is orthorhombic with 12 Fe and 4 C.
# basis are defined in this order
lattice custom 1.0 &
a1 ${acem} 0.0 0.0 &
a2 0.0 ${bcem} 0.0 &
a3 0.0 0.0 ${ccem} &
basis 0.837120 0.036661 0.250000 &
basis 0.337120 0.463339 0.750000 &
basis 0.662880 0.536661 0.250000 &
basis 0.162880 0.963339 0.750000 &
basis 0.332101 0.177370 0.068374 &
basis 0.832101 0.322630 0.931626 &
basis 0.167899 0.677370 0.431626 &
basis 0.667899 0.822630 0.568374 &
basis 0.667899 0.822630 0.931626 &
basis 0.167899 0.677370 0.068374 &
basis 0.832101 0.322630 0.568374 &
basis 0.332101 0.177370 0.431626 &
basis 0.439068 0.876994 0.250000 &
basis 0.939068 0.623006 0.750000 &
basis 0.060932 0.376994 0.250000 &
basis 0.560932 0.123006 0.750000
region cementite block 0 ${ncellxcem} 0 ${ncellycem} 4 8 units lattice
create_atoms 1 region cementite &
basis 1 1 &
basis 2 1 &
basis 3 1 &
basis 4 1 &
basis 5 1 &
basis 6 1 &
basis 7 1 &
basis 8 1 &
basis 9 1 &
basis 10 1 &
basis 11 1 &
basis 12 1 &
basis 13 2 &
basis 14 2 &
basis 15 2 &
basis 16 2
# We move a bit the cementite so it is close to the lower Fe layer.
group grcement region cementite
displace_atoms grcement move 0. 0. 2.5 units box
# Fe layer
lattice bcc 2.8553 orient x 1 -1 0 orient y 1 1 1 orient z -1 -1 2 origin 0.0 0.0 0.0 spacing ${sqrt2} ${sqrt3} ${sqrt6}
region lowerFe block 0 ${ncellxFe} 0 ${ncellyFe} 0 4 units lattice
create_atoms 1 region lowerFe
write_dump all custom coord_atoms_before_equilibrium.xyz type x y z
# Equilibration
variable temp equal 300.
variable seed1 equal 3498
variable seed2 equal 32564
velocity all create ${temp} ${seed1} mom yes dist gaussian
fix equilibrium all langevin ${temp} ${temp} 0.1 ${seed2}
fix 3 all nve
thermo_style custom step atoms time dt temp press etotal lx ly lz
thermo 100
reset_timestep 0
fix adaptdt all dt/reset 1 NULL NULL 0.1 units box
run 1000
write_dump all custom coord_atoms_equilibrium.xyz type x y z
-
As you can see, to equilibrate the system, I first set the velocities of the atoms and then used a fix langevin + nve.
The temperature reaches and fluctuates around 300K and the total energy reaches a plateau value. However, I would like to know if this is correct or if should I use in addition a minimize step before.
Would a npt fix be more appropriate? -
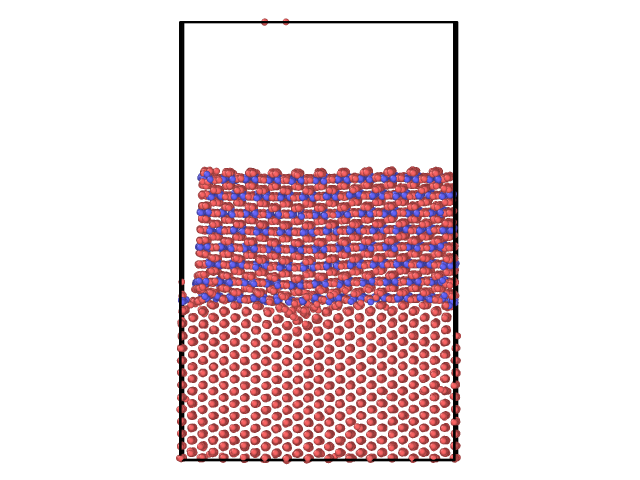
I am also concerned with the mismatch between the Fe and the cementite. Due to a difference between the lattice params of Fe and cementite, I searched for an integer number of cells in x and y such as the length of Fe is approx that of cementite (within 5%). However, the length of the cementite is a bit smaller (see picture). It is not possible to find exactly an integer number a cells such as it is exactly the same length. Since we would like to investigate the interface, I am wondering how this can affect it and if it does, how can it be solved.
-
Finally, I am also concerned by the periodic conditions that we are using in all directions. In z, we left a large empty space above the cementite in order to mimic an open surface so that Fe atoms do not interact with themselves through the PBC. However, in xy where we also use the PBC, we can see some interactions between the atoms occurs. Can this be avoided or minimized?
I leave some snapshot in order to show the different cases.
Many thanks in advance for any help you could bring me.
Christophe
PS: The LAMMPS version I am using 23 June 2022