Hi all,
I notice some non-physical behavior in my DEM jamming simulation and I wonder if it had anything to do with the bin size. For reference, my granular system (surrounded by periodic walls) is shown below and I compress it beyond the RCP volume fraction by using fix deform to shrink in the direction along the red and green plane (x and y). The top and bottom (z) along the direction perpendicular to compression is just periodic.
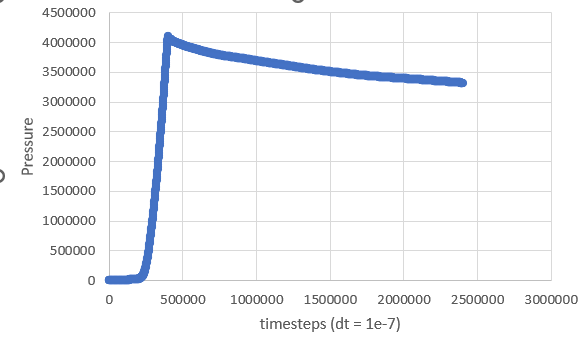
When compressed beyond the RCP threshold, I get a very high pressure, but the kinetic energy does not dissipate even with a tangential friction contact when let to equilibrate. I instead get oscillatory behavior that I don’t have an explanation for… I expect friction to dissipate this energy and the granular velocities to eventually die down to very low values (Kinetic energy of individual granules ~ < 1e-10J). When I use viscous damping, the kinetic energy momentarily goes down but shoots back up when viscosity is removed so it doesn’t help.
I wonder if it has anything to do with the number of bins… My granules have radii between 0.0045m and 0.005m and the length of the channel is 0.05m x 0.05m x 1m, therefore there are slightly more than 5 granules per length of the box in x and y direction, and 100 granules in the z-direction.
I get the following bins: binsize = 0.0149999 → bins = 4 4 67 when I use neighbor 0.02 bin. I noticed that I get the following info:
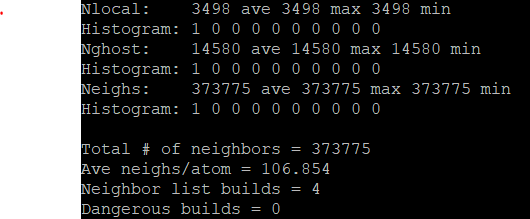
The ave neighs/atom is really large and it should ideally be close to 6 and not 106… But when I don’t use neighbor 0.02 bin, I get a smaller binsize and more bins, and a more realistic neighs/atoms of 5.3:
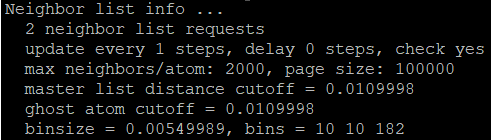
If I build the neighbor list regularly, say like 10 timesteps, will the inclusion of the neighbor command just mean more computation time but would give the same result? Also, how does binsize affect in a serial run?
At the end of a sufficiently large timestep for the case with neighbor command, I get total kinetic energy as 7.0970525J and when its not being used I get 7.095222J so it looks like I get nominally the same result. I am worried if I’m not there are some force pairs that are going undetected resulting in the lack of decrease of kinetic energy?
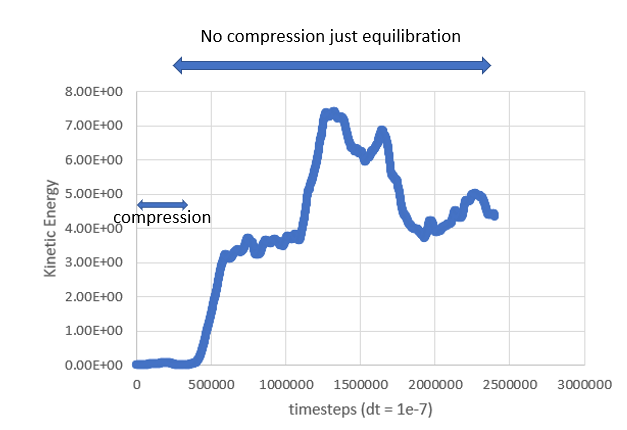
I’ve attached the kinetic energy and pressure as a function of timesteps to show what I get but I don’t reach near 0 kinetic energy even after playing around with simulation parameters
My input file is as follows, and I’ve attached my data file:
units si
atom_style sphere
boundary pp pp pp
newton off
comm_modify vel yes
read_data n_0p63_periodic_mid.sim
[n_0p63_periodic_mid.sim|attachment](upload://uiyoCBKz1QPQVOquHk0itnLhBWJ.sim) (934.1 KB)
[n_0p63_periodic_mid.sim|attachment](upload://uiyoCBKz1QPQVOquHk0itnLhBWJ.sim) (934.1 KB)
log n_0p63_periodic.lammps
#neighbor 0.02 bin
neigh_modify delay 0
pair_style gran/hertz/history 8.424908425E+10 NULL 0 0 0.05 0
pair_coeff * *
timestep 1e-7
fix 1 all nve/sphere
fix 11 all viscous 5
region atom_region block 0 0.05 0 0.05 0 1
region atom_region2 block 0.005 0.045 0.005 0.045 0.05 0.8
group atoms region atom_region
group atoms2 region atom_region2
compute peratom atoms stress/atom NULL pair
compute p atoms reduce sum c_peratom[1] c_peratom[2] c_peratom[3]
compute p1 atoms reduce sum c_peratom[4] c_peratom[5] c_peratom[6]
variable press equal -(c_p[1]+c_p[2]+c_p[3])/(3*0.05*0.05*1)#((y[896]-1e+5)-(y[895]+1e+5))*((z[893]-1e+5)-(z[894]+1e+5)))
variable stress_ratio equal c_p[1]/c_p[2]
compute peratom2 atoms2 stress/atom NULL pair
compute p2 atoms2 reduce sum c_peratom2[1] c_peratom2[2] c_peratom2[3]
compute p12 atoms2 reduce sum c_peratom2[4] c_peratom2[5] c_peratom2[6]
variable press2 equal -(c_p2[1]+c_p2[2]+c_p2[3])/(3*0.04*0.04*0.75)#((y[896]-1e+5)-(y[895]+1e+5))*((z[893]-1e+5)-(z[894]+1e+5)))
variable stress_ratio2 equal c_p2[1]/c_p2[2]
compute rot_kin all erotate/sphere # rotational kinetic energy of the spherical particles
variable myvfrac equal 0.001575126176137/vol#0.005040275060683/vol
thermo_style custom step atoms ke c_rot_kin v_press v_press2 c_p[1] c_p[2] c_p[3] c_p2[1] c_p2[2] c_p2[3] vol v_myvfrac
thermo 1000
thermo_modify lost ignore norm no # atom number non-constant
dump myDump all atom 10000 n_0p63_periodic.atom
run 200000 # final longer run
unfix 11
fix 111 all viscous 0
run 0
fix 1111 all deform 1 x vel -0.01 y vel -0.01
run 400000
unfix 1111
run 2000000
write_data n_0p63_periodic.sim