Dear all
I’am trying to calculate the electronic transport properties of oxides,
referring to Boltztrap2 example (see below link)
2019-01-04-How to use Boltztra2 interface - matgenb
In the interpolator stage, I get the following error.(see below image)
I would like to ask for any advice on this problem.
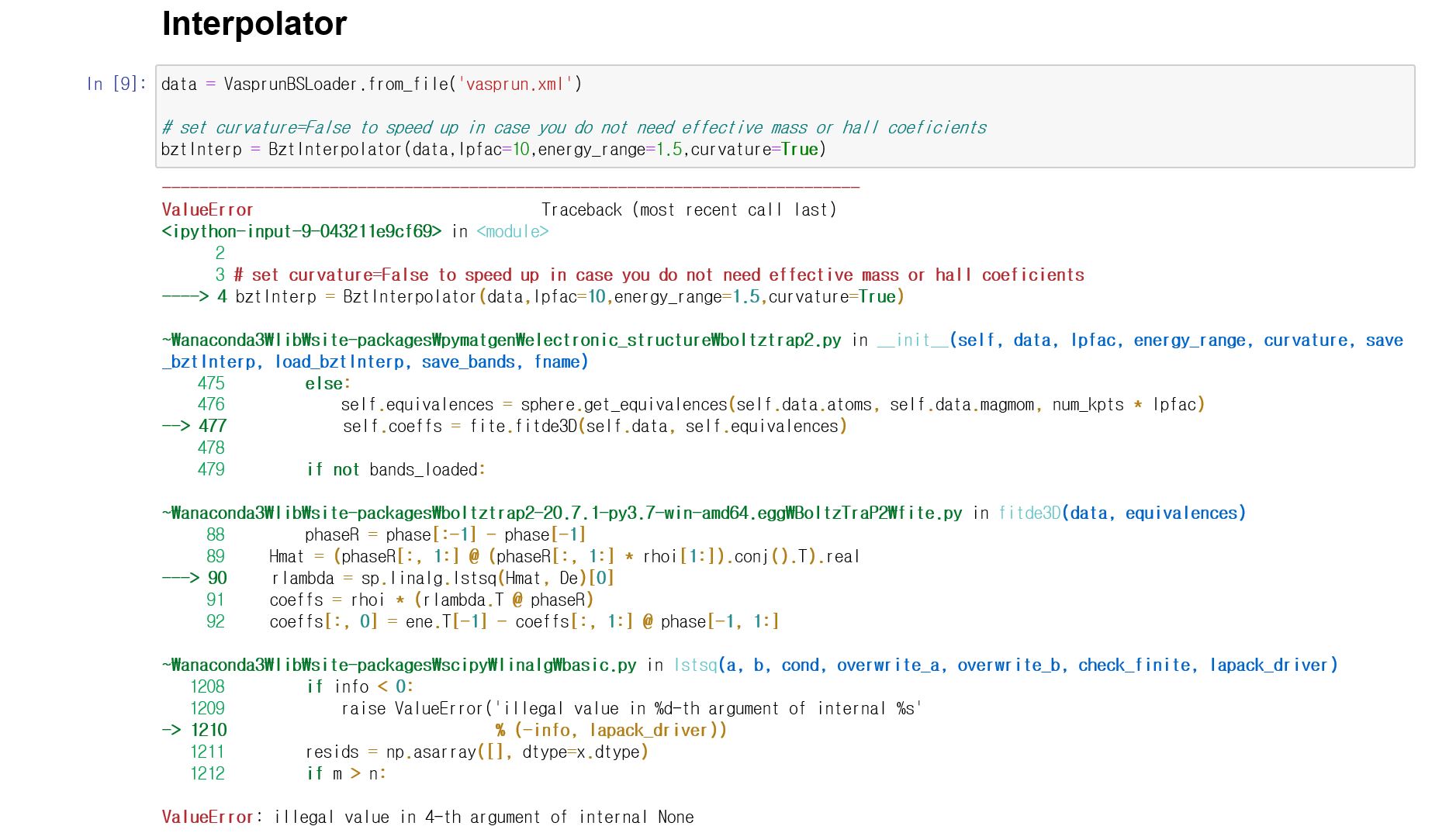
Thanks you
---------------------Computational details---------------------
- Software: VASP
-SYSTEM: mp-17844(SnWO4)
-KPOINTS
pymatgen v2021.2.14 with grid density = 1345 / number of atoms
0
Gamma
15 15 15 - INCAR
ALGO = Normal
EDIFF = 1e-05
EDIFFG = -0.01
ENCUT = 520
IBRION = 2
ISIF = 3
ISMEAR = 0
ISPIN = 2
LASPH = True
LDAU = True
LDAUJ = 0 0 0
LDAUL = 0 2 0
LDAUPRINT = 1
LDAUTYPE = 2
LDAUU = 0 6.2 0
LMAXMIX = 6
LORBIT = 11
LREAL = Auto
LWAVE = False
MAGMOM = 40.6 45.0 16*0.6
NCORE = 16
NELM = 300
NELMIN = 6
NSW = 200
PREC = Accurate
SIGMA = 0.05