Hi there,
I am encountering a “bond atom missing” error when using the bond/react command for a reversible reaction. I will describe my problem in as much detail as possible. Any help on the issues would be greatly appreciated!
As shown in the diagram below, there are two structures, A and B. I want to achieve a probability-based polymerization reaction between A and B within a specific distance range, leading to the removal of a water molecule. Please find my input file attached below.
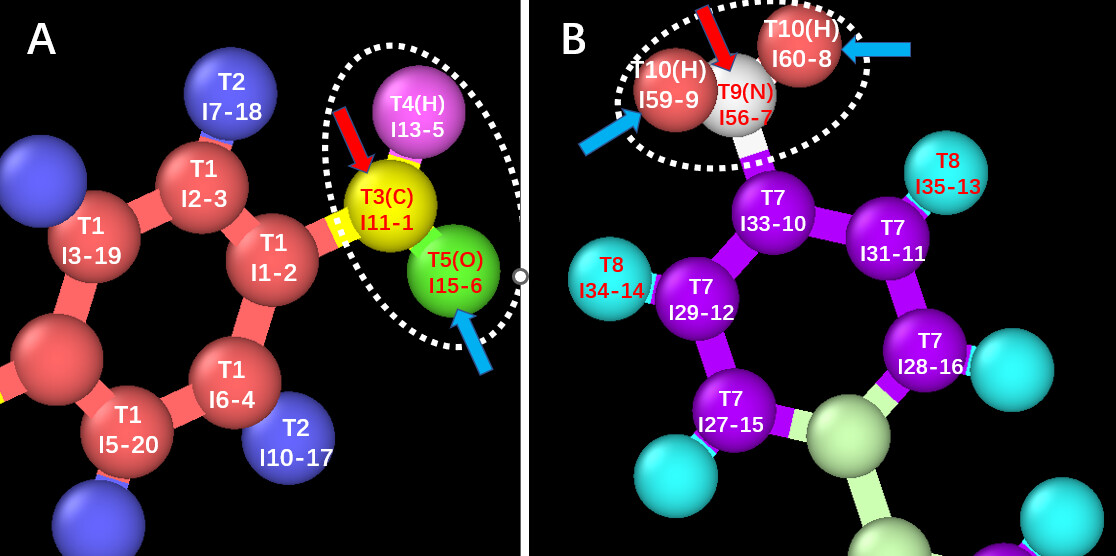
My pre and post template rules are as follows:
The T3(C)I11-1 atom and the T9(N)I56-7 atom initially form a bond. At the same time, the T5(O)I15-6 atom breaks its bond with the T3(C)I11-1 atom, and the T10(H)I59-9 atom and the T10(H)I60-8 atom each break their bonds with the T9(N)I56-7 atom. The T5(O)I15-6 atom then forms a bond with both the T10(H)I59-9 atom and the T10(H)I60-8 atom, resulting in the formation of a water molecule.
I have completed the writing of the pre and post templates and have successfully implemented the desired functionality. Following the instructions in the manual, I swapped the positions of the pre and post templates to enable reversible reactions. While the reversible reactions now occur as expected, there is a persistent instability issue with frequent “bond atom missing” errors. As shown below:
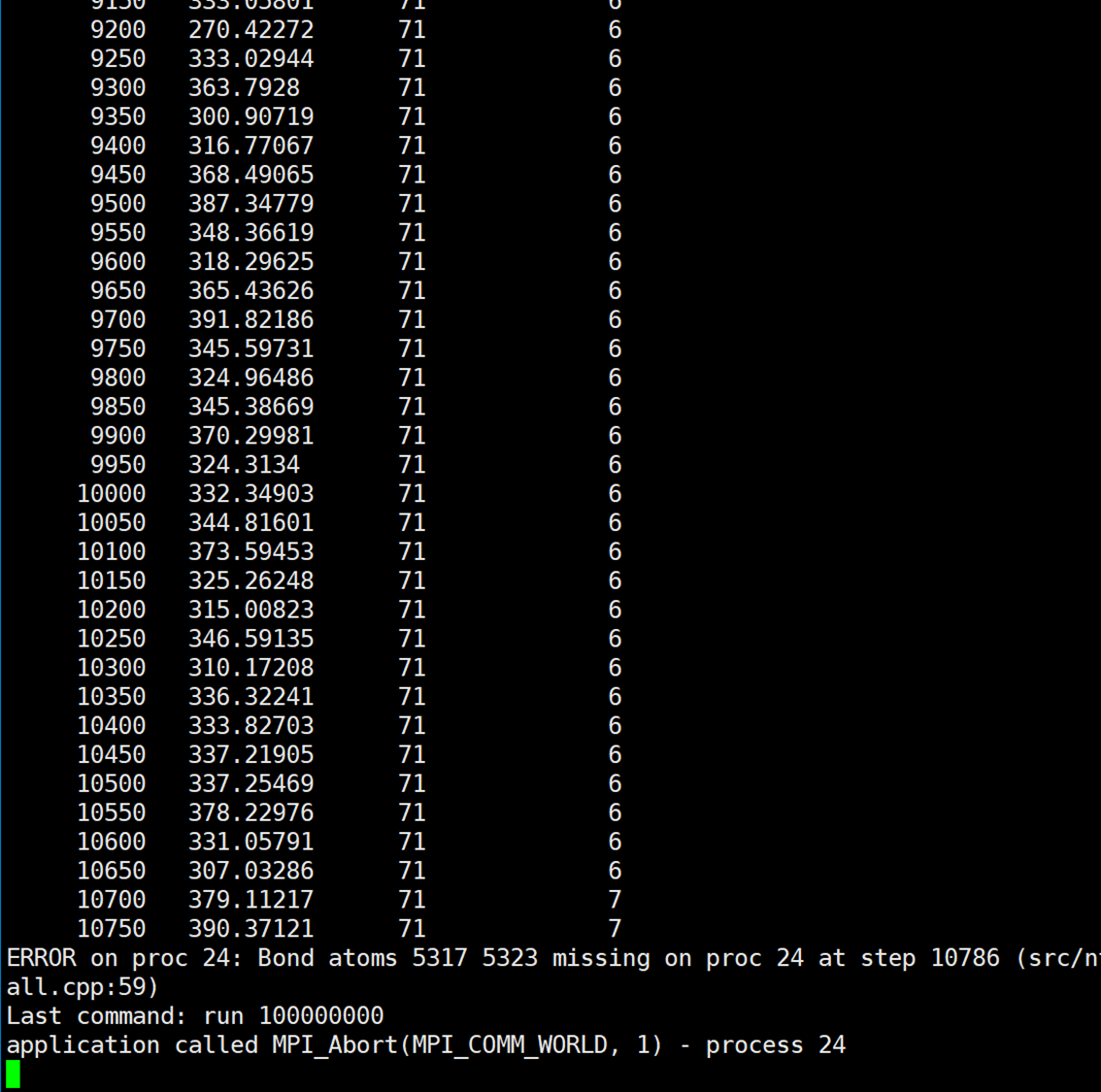
I have observed the trajectory files and found that most of the errors occur at the junction between the A monomer and the B monomer, specifically involving the T3(C)I11-1 atom and the T9(N)I56-7 atom. Additionally, there are also instances where bond atom missing errors occur with the generated water molecules.
I have tried various methods to resolve this issue. For example, I increased the Nevery value for the reverse reaction in an attempt to stagger the evaluation times of the forward and reverse reactions. I also experimented with adjusting the Rmin and Rmax values for the reverse reaction from 1 and 3 to 2 and 3 respectively. However, these attempts did not yield satisfactory results, and I am uncertain about how to proceed in resolving this problem.
Anyway, thank you all so much for your time and help.
Zilin
in.lmp (1.6 KB)
output (1.0 MB)
rxn1_stp1.map (223 Bytes)
rxn1_stp1_post.data_template (1.5 KB)
rxn1_stp1_pre.data_template (1.5 KB)
rxn1_stp1_re.map (223 Bytes)