Hello everyone,
I have created a 90% crosslinked EPON - DETDA epoxy network and i would like to perform a tensile deformation in the x axis. I also want to be able to break the bonds while the strain is applied to the network and the atoms distance is greater than the 5% of the bond length. I saw in the LAMMPS documentation that I can use the fix bond/break command. The input file I am using is the folowing:
Crosslinked-90-strain
64X32 EPON Crosslinked System with constant erate under nvt ensemble with bond breaking parameters under 5% of the bond deformation
units real
atom_style full
boundary p p p
pair_style buck/coul/long 12.0
pair_modify mix arithmetic
dielectric 1.0
special_bonds lj 0.000000 0.000000 1.000000 coul 0.000000 0.000000 1.000000
bond_style harmonic
angle_style harmonic
dihedral_style fourier
improper_style harmonic
read_data Crosslinked-90-strain.data
kspace_style pppm 0.0001
pair_coeff 1 1 15461.705185 0.323333 648.252133 # C_R
pair_coeff 1 4 5016.439576 0.286666 97.218908 # C_R H_
pair_coeff 1 5 501.643955 0.286666 9.721891 # C_R H__HB
pair_coeff 1 2 15461.705185 0.323333 648.252133 # C_R C_3
pair_coeff 1 6 19102.032807 0.315625 691.694471 # C_R N_R
pair_coeff 1 3 23260.273419 0.307916 721.983969 # C_R O_3
pair_coeff 4 4 1627.547914 0.250000 14.580000 # H_
pair_coeff 4 5 162.754791 0.250000 1.458000 # H_ H__HB
pair_coeff 4 6 6197.517816 0.278958 103.733991 # H_ N_R
pair_coeff 5 5 16.275479 0.250000 0.145800 # H__HB
pair_coeff 5 6 619.751779 0.278958 10.373399 # H__HB N_R
pair_coeff 2 2 15461.705185 0.323333 648.252133 # C_3
pair_coeff 2 4 5016.439576 0.286666 97.218908 # C_3 H_
pair_coeff 2 5 501.643955 0.286666 9.721891 # C_3 H__HB
pair_coeff 2 6 19102.032807 0.315625 691.694471 # C_3 N_R
pair_coeff 2 3 23260.273419 0.307916 721.983969 # C_3 O_3
pair_coeff 6 6 23599.444756 0.307917 738.048079 # N_R
pair_coeff 3 3 34992.280155 0.292500 804.102022 # O_3
pair_coeff 3 4 7546.629219 0.271250 108.276532 # O_3 H_
pair_coeff 3 5 754.662919 0.271250 10.827653 # O_3 H__HB
pair_coeff 3 6 28736.707926 0.300208 770.367414 # O_3 N_R
group mobile union all
timestep 1
fix 1 mobile nvt temp 298.15 298.15 10.0
velocity mobile create 298.15 1666945030 mom yes rot yes dist gaussian
fix 2 mobile deform 2000 x erate 0.0000001 remap x
#Bond Breaking
fix 3 mobile bond/break 1000 1 1.46
fix 4 mobile bond/break 1000 2 1.53
fix 5 mobile bond/break 100 1 1.07
fix 6 mobile bond/break 100 1 1.42
fix 7 mobile bond/break 100 1 1.49
fix 8 mobile bond/break 100 1 1.61
fix 9 mobile bond/break 100 1 1.14
fix 10 mobile bond/break 100 1 1.03
fix 12 mobile bond/break 100 1 1.48
fix 13 mobile bond/break 100 1 1.02
dump 1 all atom 20000 Crosslinked-90-strain.lammpstrj
dump 2 all atom 20000 Crosslinked-90-strain.dump
dump_modify 1 scale no image yes
#dump 2 all custom 20000 Crosslinked-90-strain.veldump vx vy vz
thermo_style custom step etotal ke pe ebond eangle evdwl ecoul elong temp press density lx ly lz
thermo_modify line multi
thermo 20000
thermo_modify flush yes
run 2000000
write_restart Crosslinked-90-strain.rst
wridte_data output.data
But the code keeps getting error:
error on proc 1: out of range atoms - cannot compute pppm
Does any one has any idea what I am doing wrong?
Thank you in advance all!
The “lost atoms” errors have been discussed extensively in this forum for several years. Have you tried diagnosing the initial configuration? Is it stable with this force field without deformation and bond breaking? Is it stable with deformation but without bond breaking?
Essentially, you need to show us what you’ve tried before we can really process this problem.
Maybe reduce the number of steps at which you attempt bond breaking. I guess your bonded atoms are moving too far even before you attempt to delete the bonds based on the distance criteria. You can also print the bond length information to diagnose better.
If that does not work, then it is better if you go through the previous posts on the same error as Michael pointed out, that can give you better intuition of the problem.
This whole block makes no sense. In most simulations, you would just have a single instance of fix bond/break. It most certainly makes no sense to have multiple fixes for the same bond type. What is the logic that made you write your input like this?
This kind of error often happens, when forces are too large. Please note that when you break a bond, your forces can change by quite a bit, depending on what your specific force field parameters are and what bond length you choose as cutoff. Please pay attention to the note regarding that topic in the fix bond/break documentation. Generally, you want to choose bond breaking parameters where the jump in forces is not too large.
I will pay attention to your useful advice thank you very much!
Only the two first fixes where actually in the input file. The fixes 5 -13 where commented out. There are only two types of bonds (type 1 and 2) that break simultaneously.
They were not commented out in your post. If you want useful answers, you need to make certain that you provide correct information. When you post input files, commented out lines are usually significant distractions and thus should be removed, anyway.
You are absolutely right. The correct input file is the following:
units real
atom_style full
boundary p p p
pair_style buck/coul/long 12.0
pair_modify mix arithmetic
dielectric 1.0
special_bonds lj 0.000000 0.000000 1.000000 coul 0.000000 0.000000 1.000000
bond_style harmonic
angle_style harmonic
dihedral_style fourier
improper_style harmonic
read_data Crosslinked-90-strain.data
kspace_style pppm 0.0001
pair_coeff 1 1 15461.705185 0.323333 648.252133 # C_R
pair_coeff 1 4 5016.439576 0.286666 97.218908 # C_R H_
pair_coeff 1 5 501.643955 0.286666 9.721891 # C_R H__HB
pair_coeff 1 2 15461.705185 0.323333 648.252133 # C_R C_3
pair_coeff 1 6 19102.032807 0.315625 691.694471 # C_R N_R
pair_coeff 1 3 23260.273419 0.307916 721.983969 # C_R O_3
pair_coeff 4 4 1627.547914 0.250000 14.580000 # H_
pair_coeff 4 5 162.754791 0.250000 1.458000 # H_ H__HB
pair_coeff 4 6 6197.517816 0.278958 103.733991 # H_ N_R
pair_coeff 5 5 16.275479 0.250000 0.145800 # H__HB
pair_coeff 5 6 619.751779 0.278958 10.373399 # H__HB N_R
pair_coeff 2 2 15461.705185 0.323333 648.252133 # C_3
pair_coeff 2 4 5016.439576 0.286666 97.218908 # C_3 H_
pair_coeff 2 5 501.643955 0.286666 9.721891 # C_3 H__HB
pair_coeff 2 6 19102.032807 0.315625 691.694471 # C_3 N_R
pair_coeff 2 3 23260.273419 0.307916 721.983969 # C_3 O_3
pair_coeff 6 6 23599.444756 0.307917 738.048079 # N_R
pair_coeff 3 3 34992.280155 0.292500 804.102022 # O_3
pair_coeff 3 4 7546.629219 0.271250 108.276532 # O_3 H_
pair_coeff 3 5 754.662919 0.271250 10.827653 # O_3 H__HB
pair_coeff 3 6 28736.707926 0.300208 770.367414 # O_3 N_R
group mobile union all
timestep 1
fix 1 mobile nvt temp 298.15 298.15 10.0
velocity mobile create 298.15 1666945030 mom yes rot yes dist gaussian
fix 2 mobile deform 2000 x erate 0.0000001 remap x
#Bond Breaking
fix 3 mobile bond/break 1000 1 1.46
fix 4 mobile bond/break 1000 2 1.53
dump 1 all atom 20000 Crosslinked-90-strain.lammpstrj
dump 2 all atom 20000 Crosslinked-90-strain.dump
dump_modify 1 scale no image yes
thermo_style custom step etotal ke pe ebond eangle evdwl ecoul elong temp press density lx ly lz
thermo_modify line multi
thermo 20000
thermo_modify flush yes
run 2000000
write_restart Crosslinked-90-strain.rst
wridte_data output.data
Before running this input file the system has been equilibrated in the NPT ensemble at temperature 300 for 2ns
So how much are those bonds stretched after 1000 MD steps with deformation?
How large is the change in force going to be when the bond is broken?
Imagine the process to be similar to a rope suddenly breaking under tension where the rope by default will not break and you cut it when you check. At that time the bond length may be (much?) longer than the designated cutoff distance and thus the result on the system more drastic and thus leading to atoms moving too fast and thus causing problems with PPPM.
As suggested by @Raghurame, a simple approach to verify this would be to add a compute bond/local instance to monitor bond lengths and then use compute reduce max to determine the longest bond. That will help to figure out whether checking every 1000 MD steps is sufficiently frequent.
1 Like
Thank you Axel for your helpful advice!
The initial configuration is stable with deformation. It deforms well but because there is no criteria in breaking the bonds, they remain streched for the hole duration of the run. The problem is probably the one that Axel mentions, when the bonds break there is an unstable situation in the forces and the atoms are “lost”.
I included to my code the corresponding compute commands for the bond distance and the bond forces with the commands
compute 1 mobile bond/local dist force
dump 4 all local 500 distance.data index c_1[1] c_1[2]
but when I run a constant deformation simulation with the command
fix 2 mobile deform 1000 x erate 0.0000001 remap x
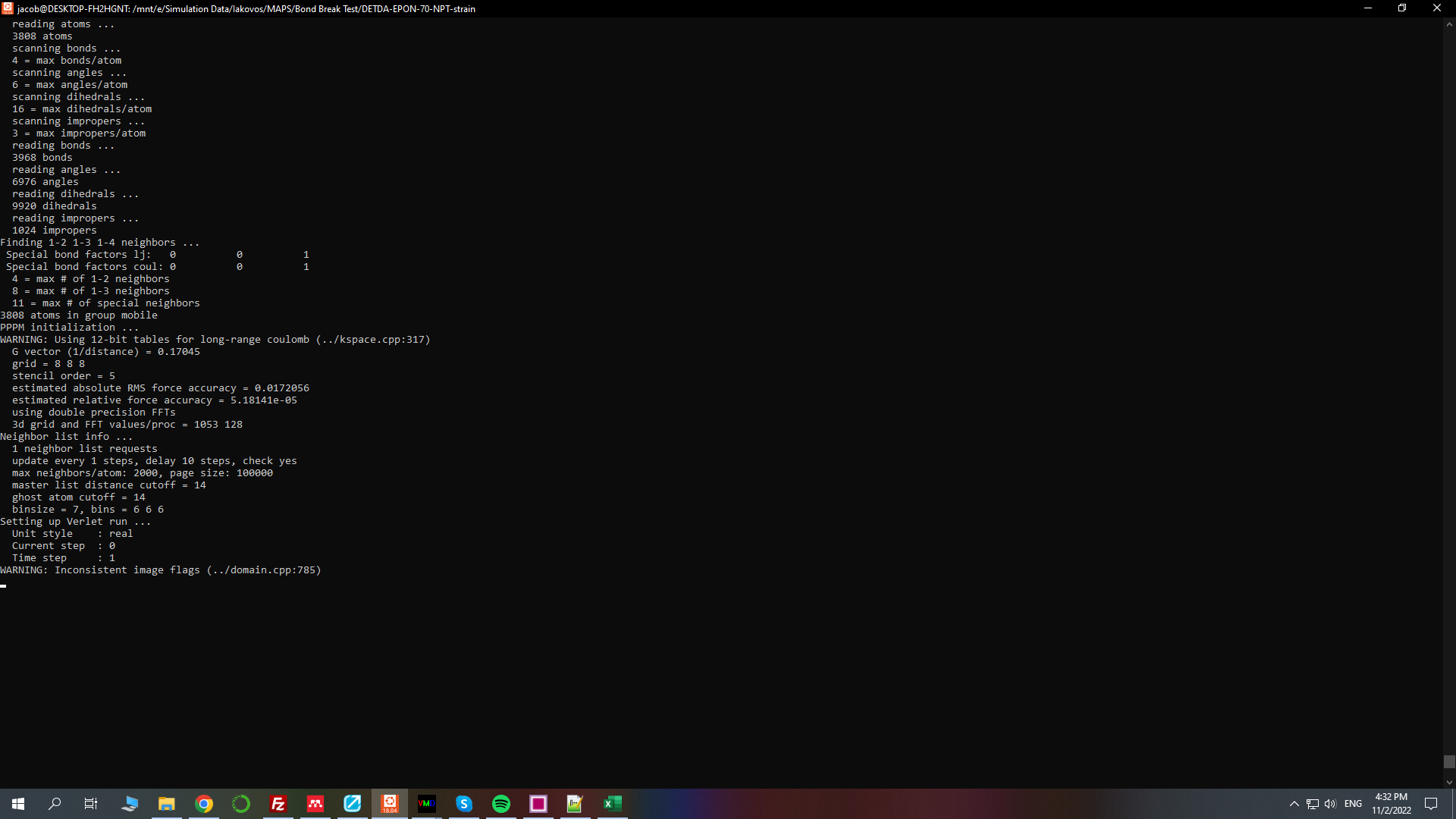
I get a warning message WARNING: Inconsistent image flags (…/domain.cpp:785)
and the simulation will not run
I upload the script to have a better look
DETDA-EPON-70-NPT-strain.in (1.9 KB)
Thank you in advance
PS: without the deformation the script runs normally
This will only remap atoms in the mobile group, yet deform the entire box. This is rarely a good idea, but certainly could be a problem.
This is a warning, not an error. The simulation will not stop because of that.
This is pretty much useless without the corresponding data file.
When I say that the simulation will not run I mean that it stops to this point
It hasn’t stopped yet. It is probably stuck in some loop possibly because of some side effect (e.g. overflow in forces) of the inconsistent image flag condition.
Ok I see, so is there any way to prevent that?
Write an input that doesn’t get your simulation into that condition.
We are reaching the point where you are wearing out my patience and are not giving me much to work with and making it needlessly difficult to help you.
What is creating a problem for you is something that can be figured out with systematic debugging on your own. What is not going to happen, that somebody from remote without the opportunity of reproducing your issues and without reliable information to begin with can solve with a simple “do this, not that” kind of advice (which seems to be what you are expecting). Setting up an advanced simulation - and that is what you are trying to do - is something that requires care and a systematic approach to adding features and checking if they are doing what they are supposed to be doing, possibly first independently and individually with simplified inputs. All the features you have using have been tested to work as documented and been applied to quite a large number of projects before. So in all probability the cause is most likely related to your system or your force field or your input.
1 Like
You are right. Thank you for your time Axel!