Hello!
I want to do some large simulations using COMB3 potential with H, C and Cu atoms.
Before that I am trying to fit some parameters (timestep and temperature damping parameter in thermostat)
and make sure that simulation copies physics more or less correctly for simple systems.
I am testing breaking bonds in simple H2 molecule. I am using Noose-Hoover thermostat, and i am heating up
molecule until the bond breaks.
At start, the distance between H atoms is 0.74 angstrom (H-H bond length).
I apply nvt fix:
fix 1 all nvt temp 1000 80000 0.05
with timestep set to 0.0005
What I expect is that the bond will break more or less close to the temperature equivalent of bond energy
(for H2 around 51k Kelvins).
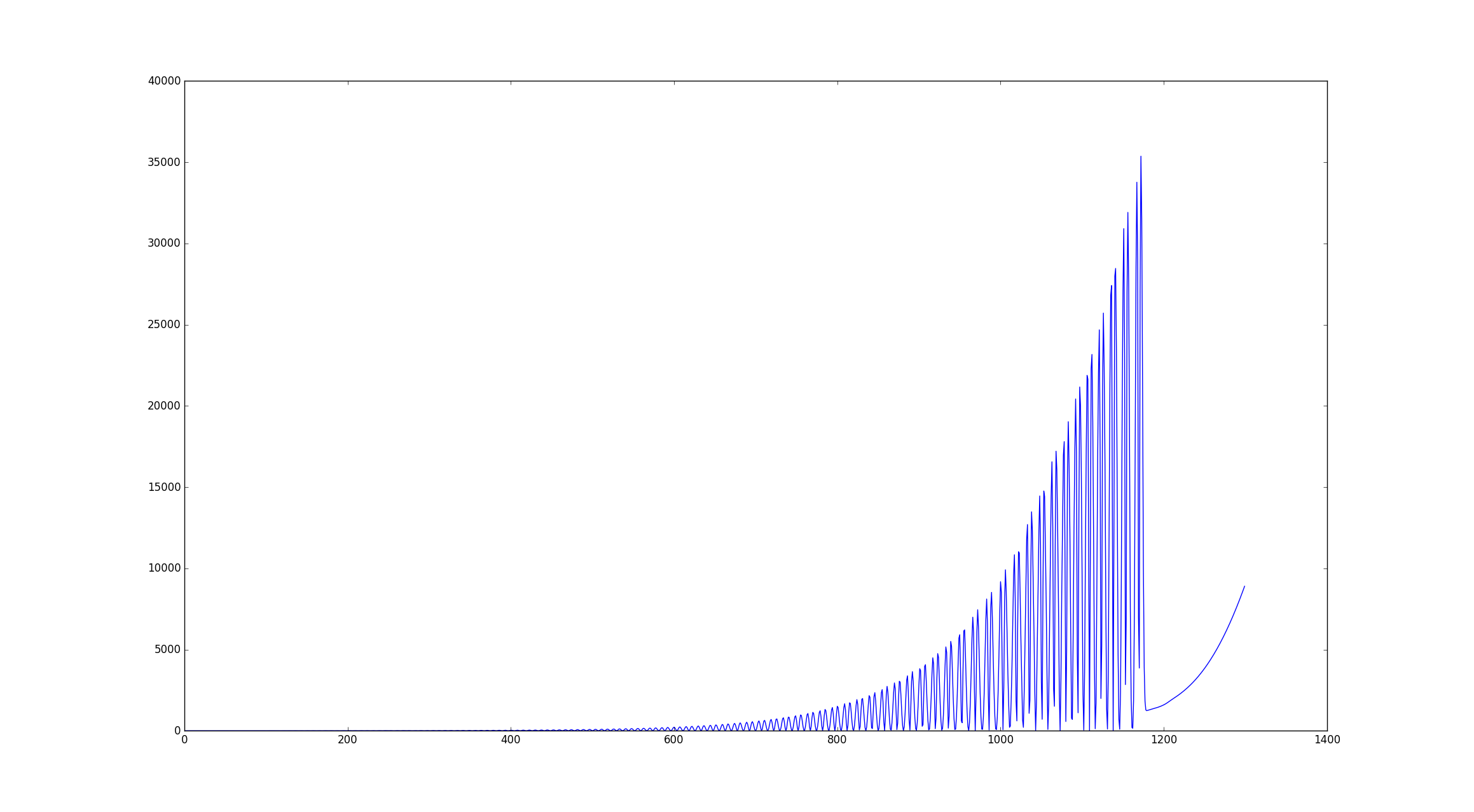
What I do to observe that the bond is broken is: visualization and observation of temperature in function of
time (attached image) and pressure in function of time.
So it is easy to see from that graph of temperature in function of time that the bond breaks at around 35k of Kelvins
because the oscillation stops.
Also in that point the pressure starts to drop dramatically.
The problem is that this is not a temperature of H2 bond break. I am fighting with all values of timestep and Tdamp
and i cannot reach that temperature. As I am not advanced Lammps user I can not understand where is the problem.
Can You, please help me and explain why it is not working as I expect it to?
Can it be a problem that I am using COMB3 for such small system with such hight temperature?
I am attaching my input and data file, and graph of temperature in function of timestep.
Best regards,
Mateusz R.
data_H2 (239 Bytes)
in.lammps_h2 (529 Bytes)
log.lammps (370 KB)
This is not so much a problem with your choice of timestep size and tdamp parameters but more likely with the COMB3 parameterization for H2 molecules. But to be fair, your timestep size is somewhat too large for systems containing H. COMB3 (like another reactive potential ReaxFF) works better with smaller timestep sizes – usually in the range of 0.1 fs to 0.2 fs. With hydrogen, you have to further reduce the timestep sizes. With high temperature simulations with H 0.05 fs (0.00005 ps) is a good number.
1 eV difference in energy is roughly 10,000 Kelvin difference in temperature. This implies that the H2 dissociation energy is roughly 2 eV (or ~200 kJ/mol) lower compared to experimental values. I will not be surprised if this is the case. COMB3 is an empirical force field fitted for bulk materials properties. Being able to qualitatively describe H2 and its dissociation (and the reaction with Cu) is a plus. To carefully characterize this, investigate the H2 dissociation energy from higher level theory of calculations – preferably coupled cluster methods – that are available from the NIST CCCBDB database (http://cccbdb.nist.gov). Compare the bond energy to COMB3 and you will get a better idea. You can vary the H2 bond distance and look at the energy variation.
Hey!
Thanks for fast reply 
I looked at that database. I have check calculated energy at 0K of reaction:
H2 -> H + H
and results for different methods vary from 36k of Kelvins to 72k of Kelvins.
I also changed the timestep as You suggested to 0.00005, and Tdamp to 0.005.
That gives me results around 47k-50k Kelvins! Thats great! I will probably try
to set it up even less to have more accurate results for my further simulations,
but i will need to check how long it will calculate.
So looking at the variation of that calculated results in database, i can accept my results
and continue creating larger system.
Thanks!
Best regards,
Mateusz Rękorajski