I have some questions regarding bond breaking in molecules with energy minimization. For this, I think it is necessary to give you short insights in what I’m trying to do with Lammps:
I am a mechanical engineer and try to modify the Lammps code for the simulation of fiber networks. For this I create a “macromolecule” which constitutes my network, where the network nodes are represented by atoms and the network connections by a modified version of the bond_style “bond”. I am then using conjugate gradient in lammps to reach a minimal energy state of the system. In the Network, it can occur that I have two bonds that are identical except for their bond coefficients, meaning that I have two bonds that connect the same atoms but have different mechanical characteristics.
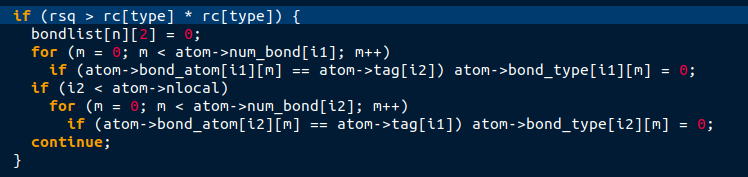
Right now, I am trying to implement bond breaking in the network. For this I use parts of the “bond_quartic” formulation. In there, the bond type of bonds meeting a certain criterion is set to 0. As I understand, this will lead to ignoring these bonds in the computation of forces and energies in subsequent iterations. I implemented this and generally got the expected results, meaning that bonds break upon meeting the specified criterion. However, whenever the special case arises, where one of the duplicate bonds breaks (duplicate as mentioned above), both bonds are disregarded in subsequent iterations, although bond breaking was only implemented for one of the two bond types. I tried figuring out where lammps checks the condition for a 0 bond type and excludes it from the list for force and energy computation. As the pointer reads as “atom->bond_type”, I thought this would happen either in the atom.cpp or the corresponding atom_vec.cpp file that I use. However, I haven’t found the corresponding lines of code. I think I am looking at the wrong files since I suspect they are only called at the beginning of the simulation. Can anybody point me to the right file and lines of code? This way I can then try to figure out what’s going on.
My second question addresses the “fix bond/break” command. As I take it, this fix only works for time integration and not for energy minimization with conjugate gradient in lammps. If I wanted to reach a formulation, in which the bonds break only after a converged step and not on every iteration. Would it be a feasible way to generate a new fix command derived from the “fix bond/break” with the corresponding “min_post_force()” method? Does such a fix already exist for energy minimization?
I cannot reproduce this. Please find below a simple example that does not require any modifications to the source code. The example creates 3 atoms and 4 bonds. Each bond
has a different type (so it could have different parameters) and there are 2 bonds defined each for the same pair of atoms.
Now one bond is disabled (the first bond i.e. bond type 1). As you can see the bond energy is correspondingly lowered. Then a second bond is disabled and all disabled bonds are removed. You can see the energy going down again, but now also the bond is no longer in the topology, which can be seen from comparing the data files.
Please note that disabling a bond is usually done by setting its type to negative, but that should not make a difference when computing interactions. The negative bond type is merely so the disabled bond can be re-enabled. With bond type 0 the removal is permanent, but just not yet from the topology.
Thus I suspect that you may have made modifications that have side effects.
units real
boundary f f f
region box block -10 10 -10 10 -10 10
atom_style bond
create_box 1 box bond/types 4 extra/bond/per/atom 4 extra/special/per/atom 4
pair_style zero 9.0
pair_coeff * *
mass * 1.0
create_atoms 1 single 0.0 0.0 0.0
create_atoms 1 single -2.0 0.0 0.0
create_atoms 1 single 2.0 0.0 0.0
bond_style harmonic
bond_coeff * 1.0 1.0
create_bonds single/bond 1 1 2
create_bonds single/bond 2 1 2
create_bonds single/bond 3 1 3
create_bonds single/bond 4 1 3
run 0 post no
write_data all.data
delete_bonds all bond 1
run 0 post no
write_data minus-1.data
delete_bonds all bond 2 remove
run 0 post no
write_data minus-2.data
Depending on your goal here, “fix bond/react” may be able to help you. If you just trying to stably relax the high-energy configurations that show up during bond breaking, “fix bond/react” has a built-in feature that locally stabilizes these types of reacting configurations.
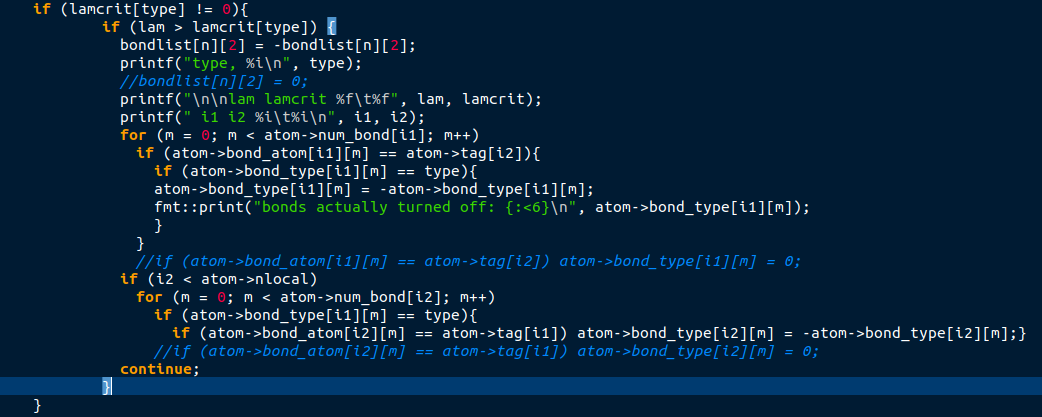
@akohlmey: Thank you for your effort. I needed some time to find my mistake and your example helped a lot by showing how the expected outputs should look like. Indeed, the mistake was in my adapted code and is fixed now.
I think that this mistake should also show up when the bond style is set to bond_quartic and two identical bonds are defined. as the code deleting bonds in the compute method of the bond style bond_quartic looks like this:
The criterions for bond breaking are different, but the important thing is the checking if (atom->bond_type == type). However, the condition of duplicate bonds might not arise when using the bond type bond_quartic. Anyways, I thought i’d post this for the sake of completeness.
@jrgissing: Thank you for your comment. I will certainly have a look into the documentation of “fix bond/react” to see if it fits my needs.
Bond style quartic in its current form is compatible only with a specific use case and for that there are no multiply defined bond between the same atoms. In fact that is a very unusual situation in general.