Hi,
Recently, I am doing some calculation with pair_style born-coul-long.
In LAMMPS, the form of born is ![]() , in front of D, the sign is plus. And in the examples of LAMMPS manual, “pair_coeff 1 1 6.08 0.317 2.340 24.18 11.51”, the D coefficient is given as 11.51, a positive number. I have analyzed that the parameters in this example are for NaCl systems with “real” units.
, in front of D, the sign is plus. And in the examples of LAMMPS manual, “pair_coeff 1 1 6.08 0.317 2.340 24.18 11.51”, the D coefficient is given as 11.51, a positive number. I have analyzed that the parameters in this example are for NaCl systems with “real” units.
So, in LAMMPS, the sign in front of D is plus, and D is set as positive for NaCl.
Well, in the work of Fusi and Tomi (M. P. Tosi and F. G. Fumi, J. Phys. Chem. Solids, 1964, 25, 31) the potential form is:
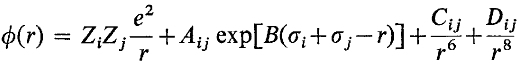
and the corresponding parameters for molten NaCl are as follows:
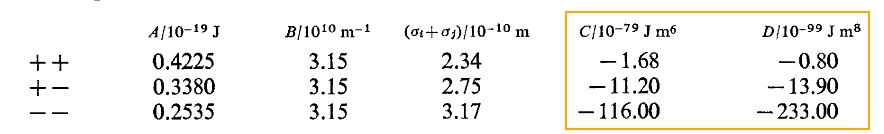
i.e. in the reference, the sign in front of D is plus, but the D is negative. The reference is also cited by LAMMPS. In fact, in many other references, the sign in front of D is minus and the D is positive.
So I think that there is some contradiction between LAMMPS and the most references, right?
What’s more, when I calculate molten NaCl, if i set the parameters with metal units as follows (D is positive), I can do the calculation and reproduce the values of reference.
pair_style born/coul/long 10.0 8.0
pair_coeff 1 1 0.2640625 0.317 2.340 1.05 0.5
pair_coeff 1 2 0.21125 0.317 2.755 7 8.6875
pair_coeff 2 2 0.1584375 0.317 3.170 72.5 145.625
kspace_style ewald 1.0e-5
But if I set D as negative, thus the net effect of the last term is subtract the 8-order dispersion interaction, which is consistent with the reference, the calculation comes up with “nan”.
I don’t know why, I am confused about it and I can’t explain it.
Would you please help me?I would appreciate your help.
Thanks very much.
Jia Wang