Dear Steve and other LAMMPS developers,
I am a first year PhD student, and one of my aspirations is to study polymer dynamics theoretically and computationally, and I plan to use LAMMPS for my simulations. I have worked with the software package before which proved to be very productive, and I plan to continue and use it for my future studies.
I have one general LAMMPS Brownian Dynamics enquiry and 3 specific questions related to the fix_langevin.cpp code, which together with fix_nve (the microcanonical ensemble in which particle number volume and energy are conserved) performs Brownian dynamics.
(1)
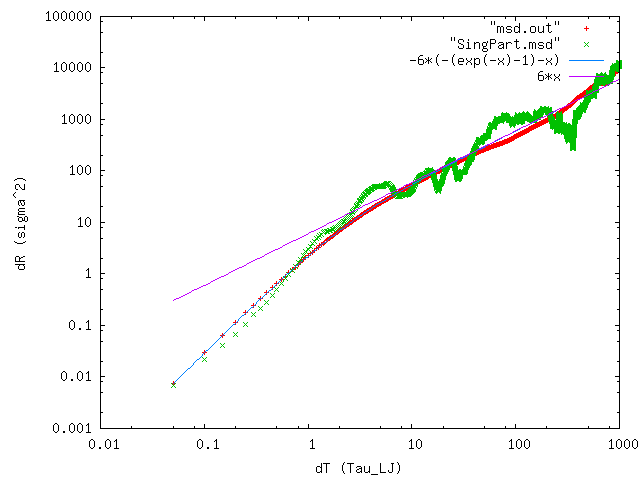
I would like to know if simulations of Brownian motion have been performed with LAMMPS such that they show the diffusion limit <x²> = Dt and fluctuations about it? I have attempted to do this with fix_nve and fix_langevin, however, when I plotted |r²| of my particle vs timestep, the behaviour was not linear as I was expecting (if needed, I can provide the script for this).
(2)
I understand that by fluctuation dissipation theorem, there exists a relation between the strength of stochastic force fluctuations and friction (hydrodynamic drag), which by equipartition can be expressed as: stochastic force intensity = 2*kBT*hydrodynamic drag.
I believe that the FDT is incorporated in the following part of the code:
for (int i = 0; i < nlocal; i++) {
if (mask[i] & groupbit) {
inertiaone = SINERTIA*radius[i]*radius[i]*rmass[i];
gamma1 = -inertiaone / t_period / ftm2v;
gamma2 = sqrt(inertiaone) * sqrt(24.0*boltz/t_period/dt/mvv2e) / ftm2v;
From documentation,
" Magnituge of stochastic force is proportional to sqrt(Kb T m / dt damp), where Kb is the Boltzmann constant, T is the desired temperature, m is the mass of the particle, dt is the timestep size, and damp is the damping factor (this is specified in time units and determines how rapidly the temperature is relaxed and is thought to be inversely related to the viscosity of the solvent). " Could someone possibly clarify the origin of this formulation as I struggle to understand how this equation is derived.
(3)
Also, I cannot quite understand what the following expressions mean, to be more precise, why are gammas assigned and multiplied as 1/ ratios:
gamma1 *= 1.0/ratio[type[i]];
gamma2 *= 1.0/sqrt(ratio[type[i]]) * tsqrt;
(4)
Having read the topics in the mailing list discussion, I understand that the prefactor (24) comes from uniform random number generator. Steve referred to Dunweg et al. when this question was raised before; having read the Dunweg paper (please find attached), I still struggle to understand where this prefactor comes from.
Thank you very much in advance!
With best wishes,
Anna Lappala
PS. Unfortunately, I cannot send the Dunweg article as an attachment to the mail-list as it is too big!