LAMMPS users,
I have been working with the Buckingham potential with coulombic tail for a little while now and I have come across a great variety of papers. Many papers use partial charges, yet some papers use the full formal charges. What advantages are there to using these partial charges as opposed to the full charges?
I also have found multiple sets of parameters for the same molecule. For instance, I have attached two excerpts from two different papers. The parameters are identical except for the O-O interactions.
I am still very new to understanding this potential - I do not understand how the same structure can be modeled with different parameters such as this. What basis is there for using one set of parameters over the other?
Thank you for your time,
Ben
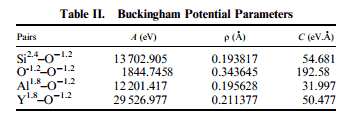
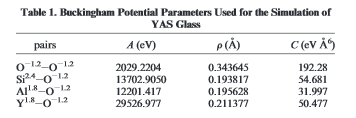
Partial charges can come out of adding the charges as a potential
fitting degree of freedom (don't forget, these are empirical
potentials).
They can be rationalized through a non-unity dielectric constant,
which means that you have screening in the material.
So is it possible to model oxides using the full or partial charge? Would you say their is an advantage of using one or the other?
Ben
So is it possible to model oxides using the full or partial charge? Would
you say their is an advantage of using one or the other?
I don't know if the 'advantage' question can be answered (at least not
by me). But besides screening the degree of ionicity of the bonds in a
particular material can be another factor influencing the choice of
charges.
These potentials are fit to a certain set of target properties and the
choice of charges will influence the quality of the fits. You'll have
to make the decision for or against using a potential based on the
fitting targets and resulting properties (i.e. transferability), not
on whether full or partial charges are used.
Daniel
There is no right/wrong, good/bad, nor advantage/disadvantage of
choosing fractional or formal charges. Charge values are just another
degree of freedom for potential developers to decide what the strength
of the Coulomb potential is. Larger charge values give you stronger
Coulomb interactions, and smaller the weaker. Therefore developers
can make their potentials more ionic or more covalent by using
different values for the charges.
As Daniel said, MD interatomic potentials are "empirical" in nature.
You fit a set of "parameters" for a certain "material" to reproduce
certain "properties". You can not miss any of the three. In your
original question, you forget the dimension of "properties". A set of
potential parameters that is fit to the "bulk elastic constants" of
rutile titania can be different from one that is fit to the "surface
energies" for the same material/phase. How to choose a set of
parameter from all existing for a certain type of material depends on
what kind of property you wish to reproduce and model.
Ray
I understand that the property in question plays a significant role. In my first post, I attached two different sets of parameters. Are you simply saying that the Oxygen parameter could potentially vary just due to the difference in properties being observed? Or could it have to do with different experimental data sets?
Ben
Yes, and more. Parameters can differ due to the following.
1) Fitted properties: what properties to fit to.
2) Target values: what values for the properties to fit to (can come
from different quantum calculations or experiments).
3) Error tolerance or weight: how well the properties are fit to.
Ray.
And here is another “hair” in the soup. The concept of partial charge has no rigorous quantum definition, i.e., partial charges are not
quantum observables. Forget about empirical potentials, in the QM world there is no operator associated to the concept of the
atoms-in-molecule (AIM) paradigm. Thus, if you were to include a review of the quantum lit you would find a ton of different ways to compute partial charges in molecular and periodic systems, Bader charges, Mulliken charges, Hirshfeld charges, ESP charges, iterative-Hirshfeld charges and so on…
Carlos