Thanks @Aaron_Kaplan for your comment.
Meanwhile, I made a quick analysis of my query cache from early 2024 vs the results of same query as of today with mp-api==0.45.1, and I see a lot of differences.
The query is the following:
mpr.materials.summary.search(
elements=["Li"],
band_gap=(0.5, 100),
energy_above_hull=(0.0, 0.05),
fields=["material_id", "formula_pretty", "nelements", "elements",
"nsites", "structure", "energy_above_hull", "volume", "band_gap"],
)
List of material IDs only in early 2024
mp-1029385, mp-1079418, mp-1079491, mp-1100430, mp-1105785, mp-1147598, mp-1173959, mp-1174205, mp-1174220, mp-1174262, mp-1174283, mp-1174285, mp-1174519, mp-1174737, mp-1176564, mp-1176626, mp-1176631, mp-1176632, mp-1176675, mp-1176702, mp-1176706, mp-1176726, mp-1177332, mp-1177369, mp-1177503, mp-1185301, mp-1192295, mp-1192707, mp-1194609, mp-1207120, mp-1208619, mp-1210869, mp-1210977, mp-1211100, mp-1218250, mp-1220728, mp-1220967, mp-1222358, mp-1222370, mp-1222452, mp-1222626, mp-1223097, mp-1223686, mp-1227900, mp-1228109, mp-1244614, mp-1247100, mp-1304584, mp-1638873, mp-1641248, mp-1666404, mp-16699, mp-18911, mp-19043, mp-23736, mp-25812, mp-25943, mp-26015, mp-26051, mp-26135, mp-26141, mp-26197, mp-26199, mp-26817, mp-29582, mp-31614, mp-555112, mp-556229, mp-558765, mp-560058, mp-606680, mp-699599, mp-704183, mp-705334, mp-705429, mp-752869, mp-753429, mp-754232, mp-754266, mp-755151, mp-755505, mp-755562, mp-756108, mp-756218, mp-756480, mp-756827, mp-757295, mp-758346, mp-758472, mp-758538, mp-759819, mp-760016, mp-760293, mp-760317, mp-766011, mp-766507, mp-766814, mp-766827, mp-767762, mp-768534, mp-768550, mp-768581, mp-768653, mp-768689, mp-770852, mp-771579, mp-772945, mp-774458, mp-775182, mp-775256, mp-777404, mp-777767, mp-778813, mp-778860, mp-779356, mp-780308, mp-780482, mp-780571, mp-8152, mp-8204, mp-849450, mp-850906, mp-861245, mp-9159, mp-942701, mp-989504, mp-989536, mp-996987, mp-998230
List of material IDs only in 2025.01.22 result
mp-1029294, mp-1029424, mp-1029517, mp-1032403, mp-1032414, mp-1034976, mp-1045540, mp-1174308, mp-1175374, mp-1176890, mp-1176904, mp-1177014, mp-1177991, mp-1196541, mp-1200046, mp-1247242, mp-1293168, mp-1304518, mp-1661789, mp-1666476, mp-19117, mp-1976674, mp-2033990, mp-2206511, mp-25397, mp-25963, mp-26105, mp-26683, mp-26846, mp-2750540, mp-2755986, mp-2761389, mp-2761607, mp-2763329, mp-2763586, mp-2763849, mp-2763899, mp-2764466, mp-2764589, mp-2766181, mp-2767670, mp-2897115, mp-2900776, mp-2911947, mp-2912291, mp-2912355, mp-504371, mp-505431, mp-532590, mp-532789, mp-540462, mp-554967, mp-555626, mp-559443, mp-561743, mp-584012, mp-585269, mp-6721, mp-672966, mp-695021, mp-696441, mp-697792, mp-723831, mp-752460, mp-752770, mp-752796, mp-752921, mp-752993, mp-753039, mp-753063, mp-753171, mp-753202, mp-753247, mp-753341, mp-753405, mp-753432, mp-753447, mp-753609, mp-753729, mp-753946, mp-754082, mp-754091, mp-754195, mp-754330, mp-754784, mp-754987, mp-755180, mp-755278, mp-755379, mp-755397, mp-755638, mp-755653, mp-755734, mp-756422, mp-756577, mp-756730, mp-756814, mp-757072, mp-757075, mp-757100, mp-757344, mp-757558, mp-757602, mp-757613, mp-757646, mp-757753, mp-757866, mp-757930, mp-758042, mp-758062, mp-758320, mp-758480, mp-758589, mp-758642, mp-758838, mp-758937, mp-759056, mp-759118, mp-759213, mp-759224, mp-759234, mp-759305, mp-759492, mp-759552, mp-759712, mp-759767, mp-759775, mp-759828, mp-759877, mp-759901, mp-760255, mp-760329, mp-760815, mp-760997, mp-761124, mp-761142, mp-761149, mp-761602, mp-761676, mp-761897, mp-762326, mp-763519, mp-763578, mp-764063, mp-764125, mp-764764, mp-764969, mp-764994, mp-765615, mp-765629, mp-766206, mp-766444, mp-766459, mp-766523, mp-767588, mp-768084, mp-768527, mp-768544, mp-768634, mp-768697, mp-768927, mp-768935, mp-768946, mp-769631, mp-769962, mp-770078, mp-770495, mp-770504, mp-770510, mp-770628, mp-770667, mp-770941, mp-770958, mp-771151, mp-771155, mp-772004, mp-772319, mp-772868, mp-773002, mp-773015, mp-773087, mp-773112, mp-773122, mp-773564, mp-774230, mp-774245, mp-774249, mp-774305, mp-774465, mp-774781, mp-774798, mp-774894, mp-774905, mp-775027, mp-775161, mp-775194, mp-775656, mp-775818, mp-775982, mp-776035, mp-776243, mp-776396, mp-776446, mp-776449, mp-776451, mp-776477, mp-776482, mp-776591, mp-776741, mp-776762, mp-777351, mp-777445, mp-777462, mp-779308, mp-779719, mp-780156, mp-780159, mp-780728, mp-781628, mp-787524, mp-850214, mp-850338, mp-850358, mp-850362, mp-850954, mp-851029, mp-865101
For the common IDs, this shows whether the values are identical for every ID:
band_gap False
elements True
energy_above_hull False
formula_pretty True
nelements True
nsites False
structure False
volume False
Here is a histogram of band gap change between the two versions for the common IDs:
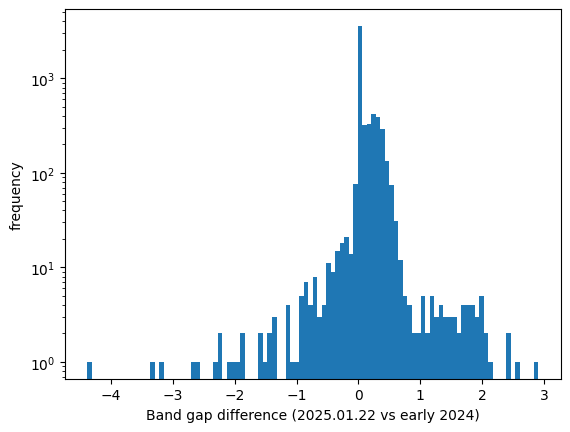
Let me know if you’d like me to share my cache from early 2024.
Hope this info helps the investigations.