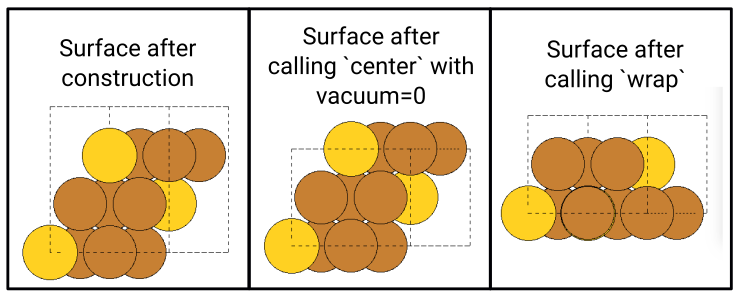
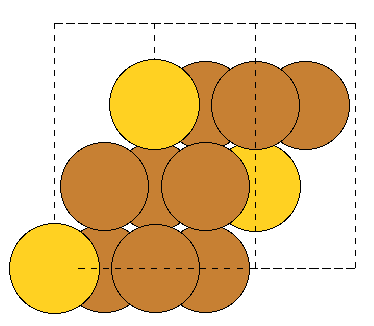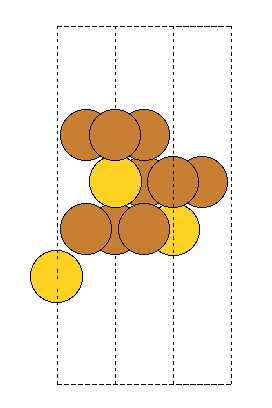Hi,
When building fcc(111) surfaces, the surface module sometimes generates wrong c-axis length. The following test code aims to generate 3 (111) layers. However, for some input fcc lattice, only 2 layers are generated (with the 3rd coinciding with the 1st layer), and hence interlayer spacing and c-axis length are wrong. The code is “““
import numpy as np
from ase import Atoms
from ase.build import bulk
from ase.build import surface
def analyze_slab(a, n_layers=3):
“”“Analyze slab created from FCC with lattice constant a”“”
# Create bulk FCC
if 1:
cu3au = Atoms('Cu3Au',
scaled_positions=[(0.5, 0.5, 0), (0.5, 0, 0.5),
(0, 0.5, 0.5), (0, 0, 0)],
cell=[a, a, a],
pbc=True)
#cu3au = bulk('Cu', 'fcc', a=a) # test primitive 1-atom FCC
# Create (111) surface
atoms = surface(cu3au, (1, 1, 1), n_layers, vacuum=0, periodic=True)
# Calculate important parameters
cell = atoms.cell
interlayer_spacing = cell[2, 2] / n_layers
in_plane_area = abs(np.cross(cell[0, :2], cell[1, :2]))
print(f"Input fcc a = {a:.6f} ")
print(f" c-axis length: {cell[2, 2]:.6f} ")
print(f" Number of layers: {n_layers}")
print(f" Interlayer spacing: {interlayer_spacing:.6f} ")
print(f" Expected interlayer spacing: {a/np.sqrt(3):.6f} ")
return atoms
for a in [3.73, 3.74,3.75,3.76,3.77,3.78,3.79,3.8]:
#print(f"Case: a = {a:.4f} Å")
atoms1 = analyze_slab(a, n_layers=3)
print()
“““. The result is:
Input fcc a = 3.730000
c-axis length: 4.307033
Number of layers: 3
Interlayer spacing: 1.435678
Expected interlayer spacing: 2.153517
Input fcc a = 3.740000
c-axis length: 6.477870
Number of layers: 3
Interlayer spacing: 2.159290
Expected interlayer spacing: 2.159290
Input fcc a = 3.750000
c-axis length: 4.330127
Number of layers: 3
Interlayer spacing: 1.443376
Expected interlayer spacing: 2.165064
Input fcc a = 3.760000
c-axis length: 6.512511
Number of layers: 3
Interlayer spacing: 2.170837
Expected interlayer spacing: 2.170837
Input fcc a = 3.770000
c-axis length: 4.353221
Number of layers: 3
Interlayer spacing: 1.451074
Expected interlayer spacing: 2.176611
Input fcc a = 3.780000
c-axis length: 4.364768
Number of layers: 3
Interlayer spacing: 1.454923
Expected interlayer spacing: 2.182384
Input fcc a = 3.790000
c-axis length: 4.376315
Number of layers: 3
Interlayer spacing: 1.458772
Expected interlayer spacing: 2.188158
Input fcc a = 3.800000
c-axis length: 6.581793
Number of layers: 3
Interlayer spacing: 2.193931
Expected interlayer spacing: 2.193931