Dear Andrew Jewett
I read the answer that you tell to Ali about ionic crystal.
I have a problem to build Al2O3 crystal.
I have built it in avogadro software and use the supper cell to create a
big crystal. but because of the unit cell is not rectangular then my
big crystal axes is not
rectangular too.
I think I us the transformation matrix to convert it but I see that
after transformation
the arrangement of atoms is not as the before.
for this I have deleted the extra atoms to modify my crystal to a
rectangular one and convert it to data file bye topotools.
But I am not sure this work is correct to delete the atoms in a crystal.
please guide me, how I can build a rectangular Al2O3 crystal.
thanks
I do not have time to write a detailed email at this time, and I am
not sure I understood your question. I'm afraid I don't know much
about the Avogadro software.
But you can always build a rectangular unit cell for a system which
has hexagonal symmetry:
For example, I have attached two different pictures to this post,
showing hexagonal and rectangular unit cells for (2-D) graphene.
However, there is no reason to use a rectangular unit cell. You can
apply rectangular boundary conditions to a hexagon-shaped molecular
object directly in LAMMPS, without needing to construct a rectangular
unit cell first.
For example, I created the following hexagonal system:
http://www.moltemplate.org/images/nanotube+water/nanotube+walls+water_top_nopbc_t=0.jpg
But when I run it in LAMMPS, I use rectangular boundary conditions:
http://www.moltemplate.org/images/nanotube+water/nanotube+walls+water_side_pbc_t=0ps.jpg
This is okay. LAMMPS has no trouble to simulate this system even
though the initial coordinates of the atoms do not lie within the
rectangular boundary conditions.
I suggest you use a program to create a a PDB file (or an XYZ file)
for your Al2O3 unit cell. It could be either a rectangular or a
hexagonal unit cell. At this point, you have two choices:
1) Load the structure in VMD, and convert it to a LAMMPS data file
using topotools. Topotools can make multiple copies of the the
unit-cell structure to create a larger crystal (supercell). (VMD's
"Solvate" plugin may be able to do this as well, by replicating the
PDB file.) Alternately, once you have created a LAMMPS data file for
your unit cell, you can convert it to an .LT file (using
"ltemplify.py"), and build a crystal out of it using moltemplate's
"new" command.
2) manually copy and paste the coordinates from the PDB file into the
"Data Atoms" section of an .LT file, and use moltemplate's "new"
command. (Perhaps I should add a read_pdb() command in moltemplate).
I have to go now. No time.
If you have time, please clarify your question.
Cheers
Andrew
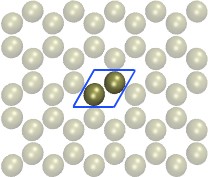
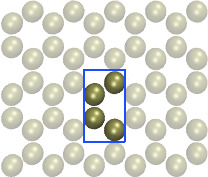
Andrew's advice about using the moltemplate tool is
all good. I'll simply add that for the 2 pictures
appended to his message, if there are no explicit
bonds between the atoms, then you can generate
those systems in LAMMPS directly for any size
you wish, using the lattice custom command. The
2nd formulation is simpler, a 4-atom unit cell and
orthogonal simulation box. But you could also
do it the first way with a 2-atom unit cell and a triclinic
simulation box.
Steve
Dear Andrew Jewett
Thanks for your attention and your answer.
The Avogadro is a software to building a molecule and crystal in pdb or xyz format.
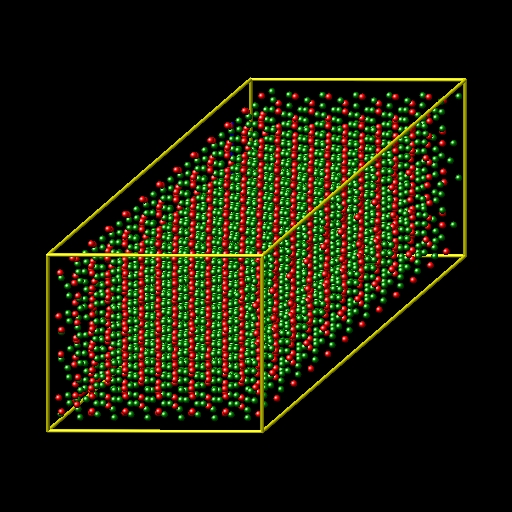
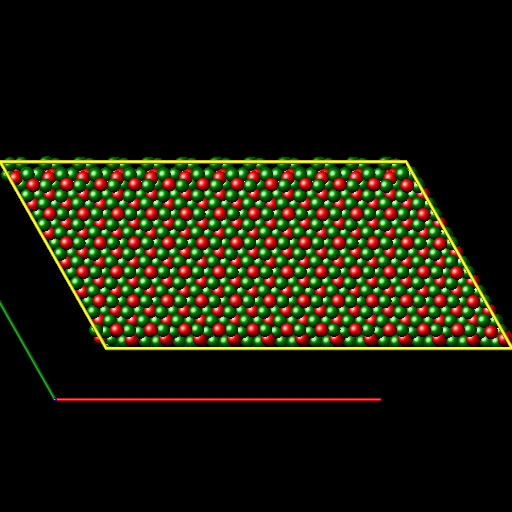
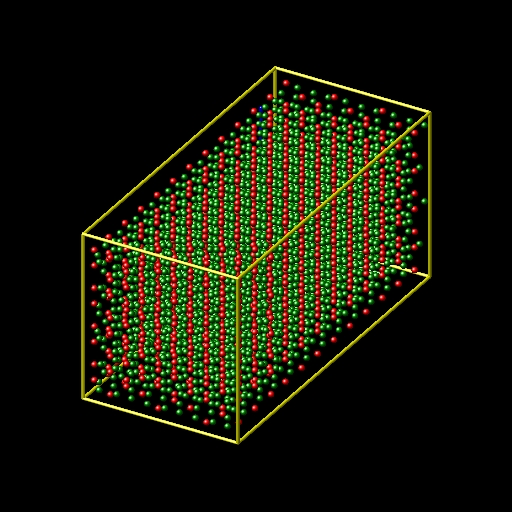
I have built the Al2O3 crystal in xyz format by replicated (supper cell) the Al2O3 conventional unit cell and then I achieve a file like pic 1 & 2.
but because of I need a cubic Al2O3 crystal, I have deleted the extra atoms of my xyz file to create the cubic crystal like pic 3.
By deleting the extra atoms, I can create a Al2O3 cubic crystal by terminated to Al atoms or to oxygen atoms, but the results of simulation (stiffness coefficient) are different for this tow cases.
for this reason, I prefer to create a cubic Al2O3 crystal without deleting the atoms.
I hope I have clarified my question.
Thanks
I'm sorry for the delay. I am very busy right now. It will get
better later in the month.
(When you say that you "deleted" the atoms, what do you mean? Did you
delete the atoms using the Avogadro software? Did you edit the XYZ or
DATA file with a text-editor?)
I don't know if the difference in the stiffness which you observed is
significant. I will assume that it is.
In any case, if there is a problem, then it would be in your data file
that you are feeding to LAMMPS. (I assume you are using a DATA file.)
You need to check this file.
Have you opened your LAMMPS data file using a text editor to verify
that the number of atoms is correct?
Have you tried visualizing your data file using VMD and topotools?
I have attached some instructions for looking at data files using VMD
and topotools.
There are instructions at the end of this file which show you how to
display the periodic boundary box, and wrap the atomic positions
inside this box. You can also check for atom overlap.
I won't be checking email very often in the next week or two.
Cheers!
Andrew
README_visualize.txt (2.85 KB)