Hi @munrojm, thanks for the updates.
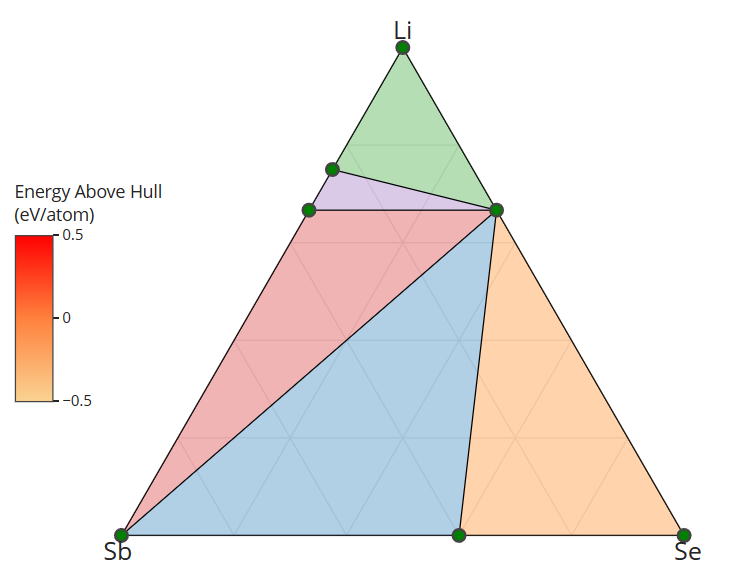
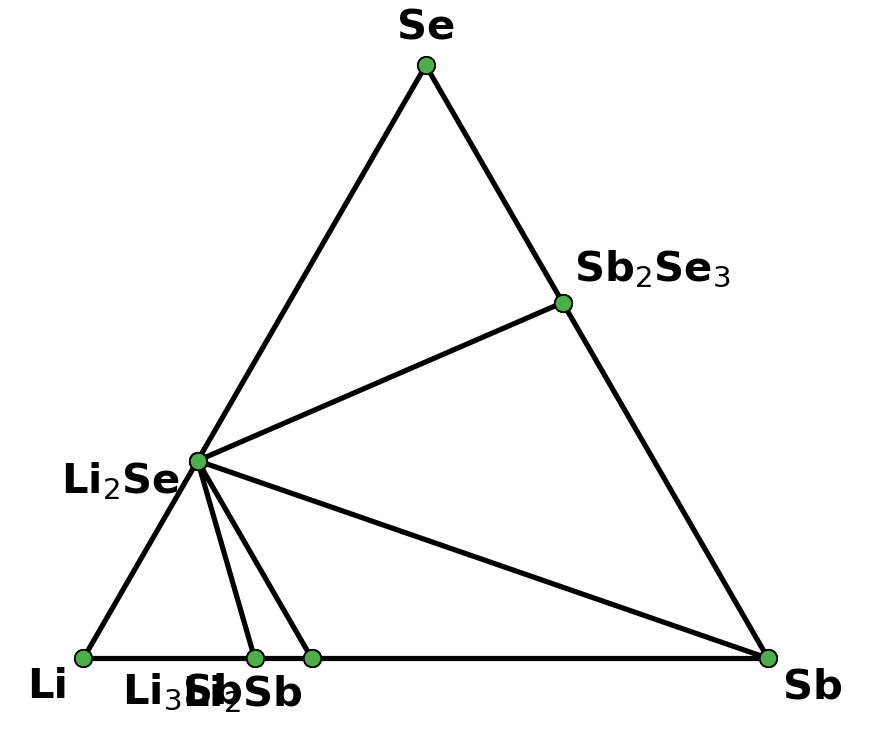
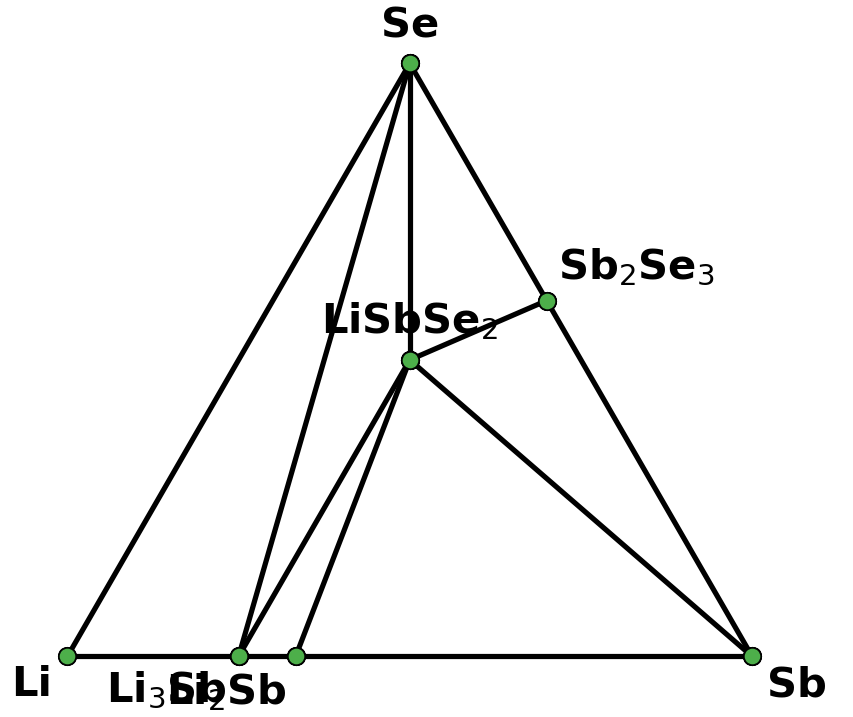
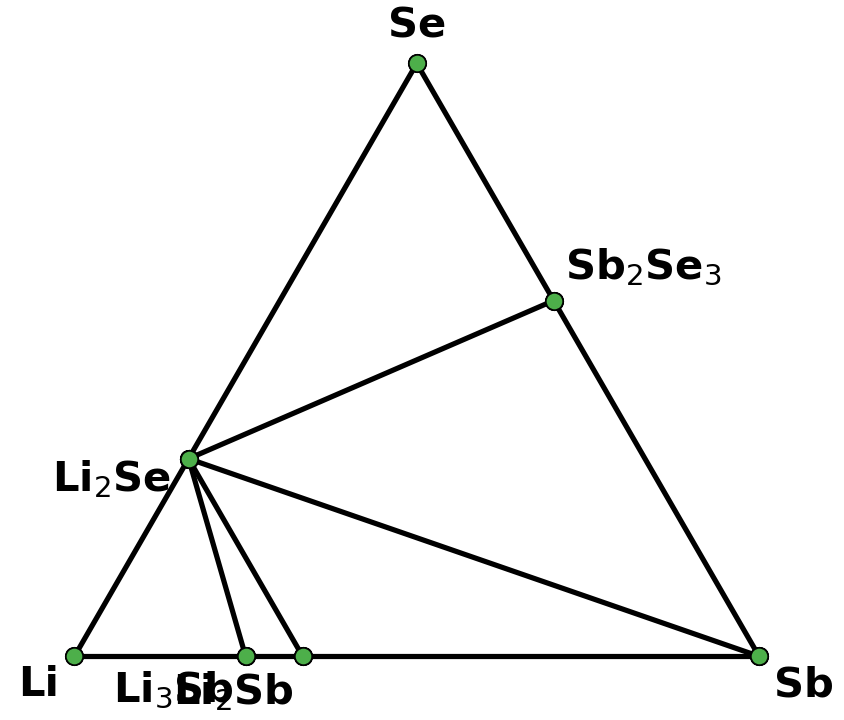
I retrieved entries to build phase diagram locally because I found that phase diagrams displayed by material project webapp are sometimes inconsistent with the self-built one before the new API and database are debuted. And I guess the webapp loads the pre-computed phase diagram, rather than always builds them by entries, right? Therefore, I think pre-computed phase diagrams are not always reliable, as they are possibly out-of-date, due to some updates like new entries, correction schemes, and database releases, etc.
entryList = mpr.get_entries_in_chemsys(‘Li-Rh-F’, additional_criteria={‘thermo_types’:[ThermoType.GGA_GGA_U_R2SCAN]})
Following your tips, I specified the thermo_type as GGA/GGA+U/R2SCAN, and got a list of entries. These entries have different tags on energy adjustments, including MP2020 anion correction, MP GGA(+U)/R2SCAN mixing adjustment, and None adjustment.
Should I confirm that the retrieved entries had been processed through the compatibility of GGA(+U)/R2SCAN mixing scheme? Or I should process them again. However, when I attempted to process them by myself:
entryList = MaterialsProjectDFTMixingScheme().process_entries(entryList, clean=True)
The error raised as below. I’d like to know how to resolve it to proceed.
warnings.warn(
Retrieving ThermoDoc documents: 100%|█████████████████████████████████████████████████| 40/40 [00:00<?, ?it/s]
Processing 16 GGA(+U) and 24 R2SCAN entries…
D:\Anaconda\envs\pymatgen2023\Lib\site-packages\pymatgen\entries\compatibility.py:1044: UserWarning: Failed to guess oxidation states for Entry mp-1185455-GGA (LiRh3). Assigning anion correction to only the most electronegative atom.
warnings.warn(
D:\Anaconda\envs\pymatgen2023\Lib\site-packages\pymatgen\entries\compatibility.py:1044: UserWarning: Failed to guess oxidation states for Entry mp-1185348-GGA (LiF3). Assigning anion correction to only the most electronegative atom.
warnings.warn(
D:\Anaconda\envs\pymatgen2023\Lib\site-packages\pymatgen\entries\compatibility.py:1044: UserWarning: Failed to guess oxidation states for Entry mp-974386-GGA (Rh3F). Assigning anion correction to only the most electronegative atom.
warnings.warn(
D:\Anaconda\envs\pymatgen2023\Lib\site-packages\pymatgen\entries\compatibility.py:1044: UserWarning: Failed to guess oxidation states for Entry mp-1209757-GGA (RhF6). Assigning anion correction to only the most electronegative atom.
warnings.warn(
Processed 16 compatible GGA(+U) entries with MaterialsProject2020Compatibility
Entries belong to the {‘Li’, ‘F’, ‘Rh’} chemical system
Generating mixing state data from provided entries.
Traceback (most recent call last):
File “D:\Seafile\peikai\My Libraries\Repos\Volume-Planning-for-Anodes\DFTMixingScheme.py”, line 18, in
entryList = MaterialsProjectDFTMixingScheme().process_entries(entryList, clean=True)
^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^
File “D:\Anaconda\envs\pymatgen2023\Lib\site-packages\pymatgen\entries\mixing_scheme.py”, line 181, in process_entries
mixing_state_data = self.get_mixing_state_data(entries_type_1 + entries_type_2, verbose=False)
^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^
File “D:\Anaconda\envs\pymatgen2023\Lib\site-packages\pymatgen\entries\mixing_scheme.py”, line 567, in get_mixing_state_data
for group in self.structure_matcher.group_structures(l_pregroup):
^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^
File “D:\Anaconda\envs\pymatgen2023\Lib\site-packages\pymatgen\analysis\structure_matcher.py”, line 848, in group_structures
inds = list(inds)
^^^^^^^^^^
File “D:\Anaconda\envs\pymatgen2023\Lib\site-packages\pymatgen\analysis\structure_matcher.py”, line 845, in
lambda i: self.fit(refs, unmatched[i][1], skip_structure_reduction=True),
^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^
File “D:\Anaconda\envs\pymatgen2023\Lib\site-packages\pymatgen\analysis\structure_matcher.py”, line 609, in fit
match = self._match(struct1, struct2, fu, s1_supercell, break_on_match=True)
^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^
File “D:\Anaconda\envs\pymatgen2023\Lib\site-packages\pymatgen\analysis\structure_matcher.py”, line 709, in _match
return self._strict_match(
^^^^^^^^^^^^^^^^^^^
File “D:\Anaconda\envs\pymatgen2023\Lib\site-packages\pymatgen\analysis\structure_matcher.py”, line 760, in strict_match
if LinearAssignment(mask).min_cost > 0: # pylint: disable=E1101
^^^^^^^^^^^^^^^^^^^^^^
File “pymatgen\optimization\linear_assignment.pyx”, line 72, in pymatgen.optimization.linear_assignment.LinearAssignment.init
File "D:\Anaconda\envs\pymatgen2023\Lib\site-packages\numpy_init.py", line 284, in getattr
raise AttributeError("module {!r} has no attribute "
AttributeError: module ‘numpy’ has no attribute ‘int’