Dear All,
I’m gonna simulate the couette flow of water with LAMMPS. I read the mailing lists about how to build the data.file and realized that there are different softwares to do this (VMD, Packmol, Moltemplate, etc).
http://lammps.sandia.gov/prepost.html
https://sites.google.com/site/akohlmey/software/topotools/topotools-tutorial—introduction
I am wondering which of those softwares is more usual and convenient to build the molecules of water between two Copper walls? Please advise.
Best regards,
Hamed.
Just a short reply.
PACKMOL is a program for generating atomic coordinates. It does not
generate LAMMPS DATA files or force-field parameters. However it
creates coordinate files which either VMD or moltemplate can read.
You still have to define bonds and force-field parameters. (I am also
not sure whether PACKMOL can generate crystalline structures.)
However both topotools(VMD) and moltemplate can generate LAMMPS DATA
files (containing bonds, etc) for water confined between two sheets:
For an example, I suggest you visit www.moltemplate.org, and scroll
down to the example named "Carbon-Nanotube Capillary". In
moltemplate, you would probably split a system like this into multiple
different files which define the different objects in your simulation
(walls, water, etc...). Then you link them together in your
"system.lt" file which refers to all of these other files. Here are
the relevant files:
http://moltemplate.org/examples/nanotube+water/system.lt
http://moltemplate.org/examples/nanotube+water/water_box.lt
http://moltemplate.org/examples/nanotube+water/spce.lt
# You would replace the next two files with copper walls instead of graphene:
http://moltemplate.org/examples/nanotube+water/graphene.lt
http://moltemplate.org/examples/nanotube+water/graphene_walls.lt
# Your system might be somewhat simpler because you don't have a nanotube.
# You would convert these files into LAMMPS format using this command:
# moltemplate.sh system.lt
# or
# moltemplate.sh system.lt -vmd
There are some alternative approaches, and suggestions here:
http://moltemplate.org/examples/nanotube+water/chiral_nanotubes.html
http://moltemplate.org/examples/nanotube+water/README_realistic_junctions.txt
If you need to connect your copper atoms together with explicit bonds,
then this is currently a little bit more difficult, but it is
possible. (In that case, please read the other email that I posted
today to the LAMMPS mailing list.)
I hope this gets you started.
Andrew
Although not as general a solution, Laurent Joly wrote a perl script
to build a similar system: "capillary filling of a CNT with water"
http://lammps.sandia.gov/user.html (scroll down)
Hello Andrew,
Thank you very much for your help and comments.
I have started simulation of water between two graphene walls (instead of copper) and changed the files of moltemplate-files in “Carbon-Nanotube Capillary” accordingly to generate the molecules of water between two plates. Considering the Couette flow example of LAMMPS, I added a few commands to define velocity for walls as follow: (In this step, I have set velocity for Cgrahene group which was defined for both top and bottom walls).
velocity mobile create 1.0 482748 temp tempMobile
velocity Cgraphene set 50.0 0.0 0.0
fix 3 Cgraphene setforce 0.0 0.0 0.0
However, when I visualize the dump.file, the walls are stationary and I don’t know why they don’t move? I was wondering if you could help me how to approach the problem? I have also attached the image of simulation result and run.in.ntv file to this email.
Best reagrds,
Hamed.
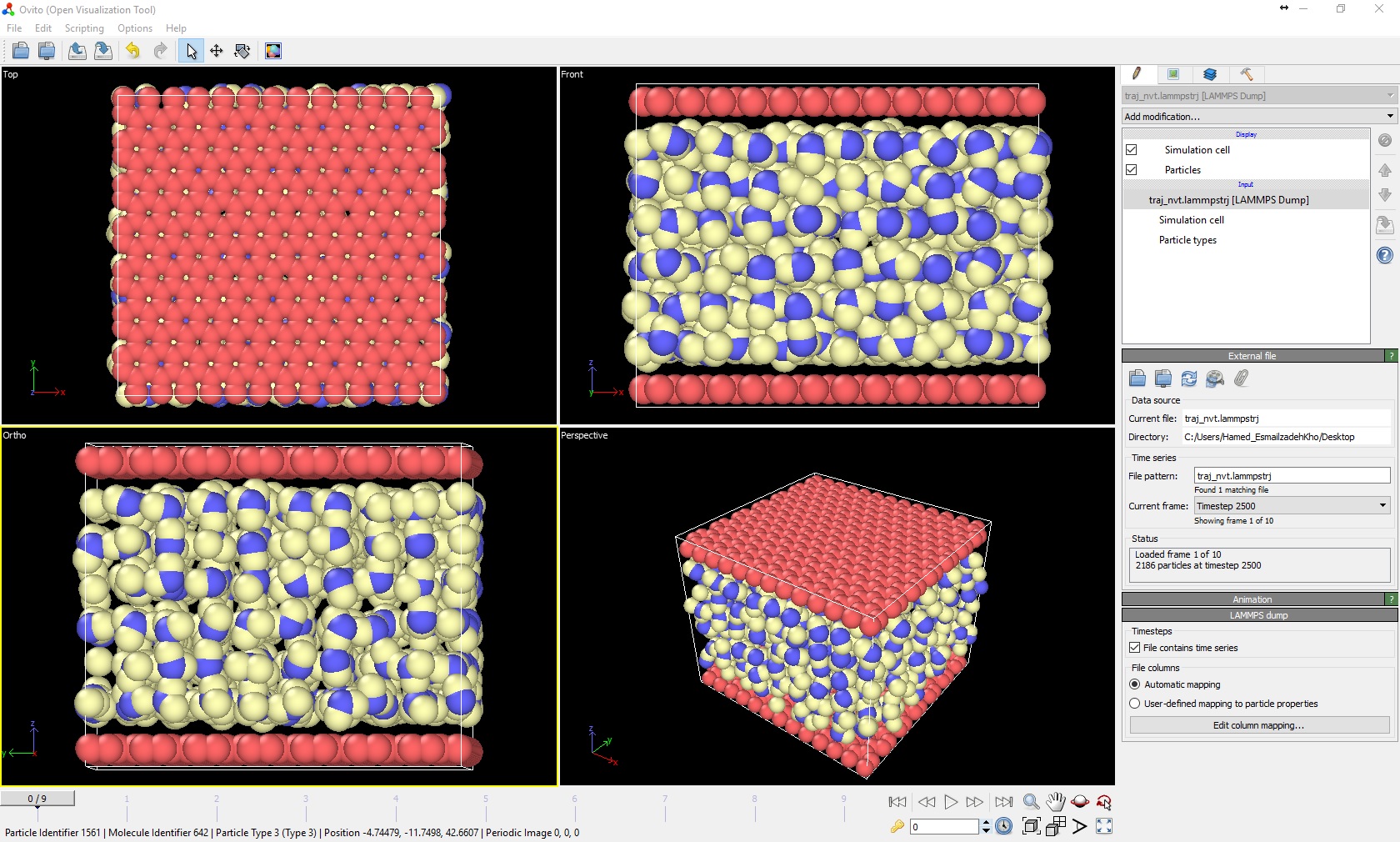
However, when I visualize the dump.file, the walls are stationary and I don’t know why they don’t move? I was wondering if you >could help me how to approach the problem?
Have you dumped the coords of the wall atoms to a dump file.
Do they change on successive timesteps? If so, it could
be a viz issue.
If they don’t change, then start commenting out things
you have added to the script that worked (in.couette)
to see what changes the behavior.
In other words, you can do some debugging to figure
how what is going wrong with your input script.
Steve
Hello Andrew,
Thank you very much for your help and comments.
I have started simulation of water between two graphene walls (instead of
copper) and changed the files of moltemplate-files in "Carbon-Nanotube
Capillary" accordingly to generate the molecules of water between two
plates. Considering the Couette flow example of LAMMPS, I added a few
commands to define velocity for walls as follow: (In this step, I have set
velocity for Cgrahene group which was defined for both top and bottom
walls).
velocity mobile create 1.0 482748 temp tempMobile
velocity Cgraphene set 50.0 0.0 0.0
fix 3 Cgraphene setforce 0.0 0.0 0.0
However, when I visualize the dump.file, the walls are stationary and I
don't know why they don't move? I was wondering if you could help me how to
approach the problem? I have also attached the image of simulation result
and run.in.ntv file to this email.
i don't see a fix doing time integration of the atoms in the Cgraphene group.
without time-integration, atoms don't move.
axel.