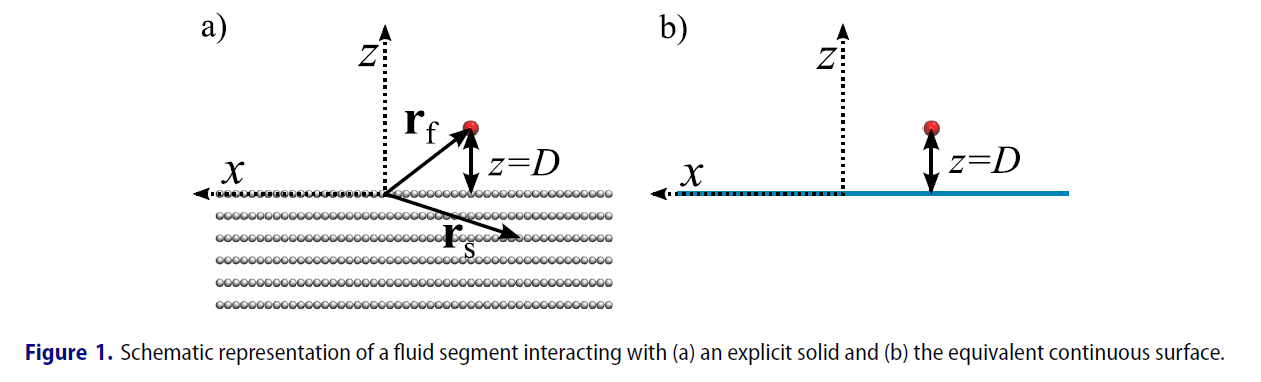
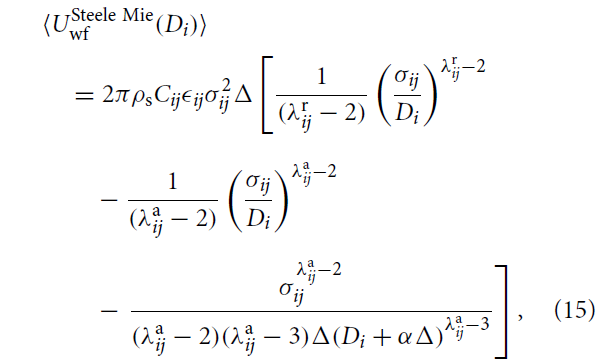I plan to calculate the solid surface-fluid energy, and compare both explicit solids and implicit solid (essentially a wall) system. The user defined potential function is a 10-4-3 steele potential function modified with Mie-potential, as a function of distance from wall in z-direction.
If it’s a function of just particle-wall distance you should be able to do it with fix addforce, taking a carefully defined per-atom variable as the z-force. You can define per-type coefficients with per-atom variables using boolean overloading:
I see… thanks for the reply. How about the pair_style table?
I read from the section we can prepare a table by using pair_python and pair_write to prepare the table with the user defined function, but I’m not sure is that possible to produce the table based on the equation 15. Then if possible, maybe I can just compute group/group between the particle and the solid surface?
This may work for explicit solid system but I think is not applicable to implicit wall…
Yes is it just a function of particle-wall distance, it this can work it will be great! Can I know more on this detail on how to create the implicit wall from fix addforce command?
''variable sigma atom “(type == 1) * 0.25 + (type == 2) * 0.69 + …”
Do you mean, for example, the wall is atom type A, particle is atom type B, then there will 3 sigma atom type , sigma A, sigma B, but how about the sigma AB?
I do not know how to calculate the parameters like \sigma_{ij} and so on. I don’t know the model you are using, and even if I did, you need to verify all calculations for yourself, since I am just a stranger on the internet
Look up what the LAMMPS manual says about fix addforce. The manual should tell you that you can set it to impose a per-atom force, that is, a variable which can take in some data about every single atom (in your case, each atom’s type and its z-coordinate) and calculate a formula that you specify.
Working backwards, if you can work out (1) what force the wall must exert on each atom given its type and location (2) how to encode that into per-atom variables, fix addforce will then impose precisely that force on each atom at each timestep – which will have achieved what you are wanting.