N.B. The problem is solved by taking co-ordinates of atom ids 6, 9 and 2049 in the final.txt file. Now I am getting 0.169 eV migration barrier just like the literature.
I am trying to calculate the interstitial migration energy of Ni by transforming a [100] dumbbell to a [010] dumbbell on {100} plane. I am creating the dumbbell by shifting a Ni atom to x or y direction by 30% of the lattice parameter. Next, I’m creating a new Ni atom that is equally spaced out from the lattice site on the opposite direction. For making both [100] and [010] configuration, I am using the perfect Ni structure and then placing the interstitial atom in it.
After creating the dumbbells, I am minimizing the both systems with the following commands:
min_style cg
minimize 1e-15 1e-15 5000 5000
These are the snapshots of the initial and the final configs (the numbers on the atoms are the corresponding atom ids, where 2049 being the newly created Ni atom) -
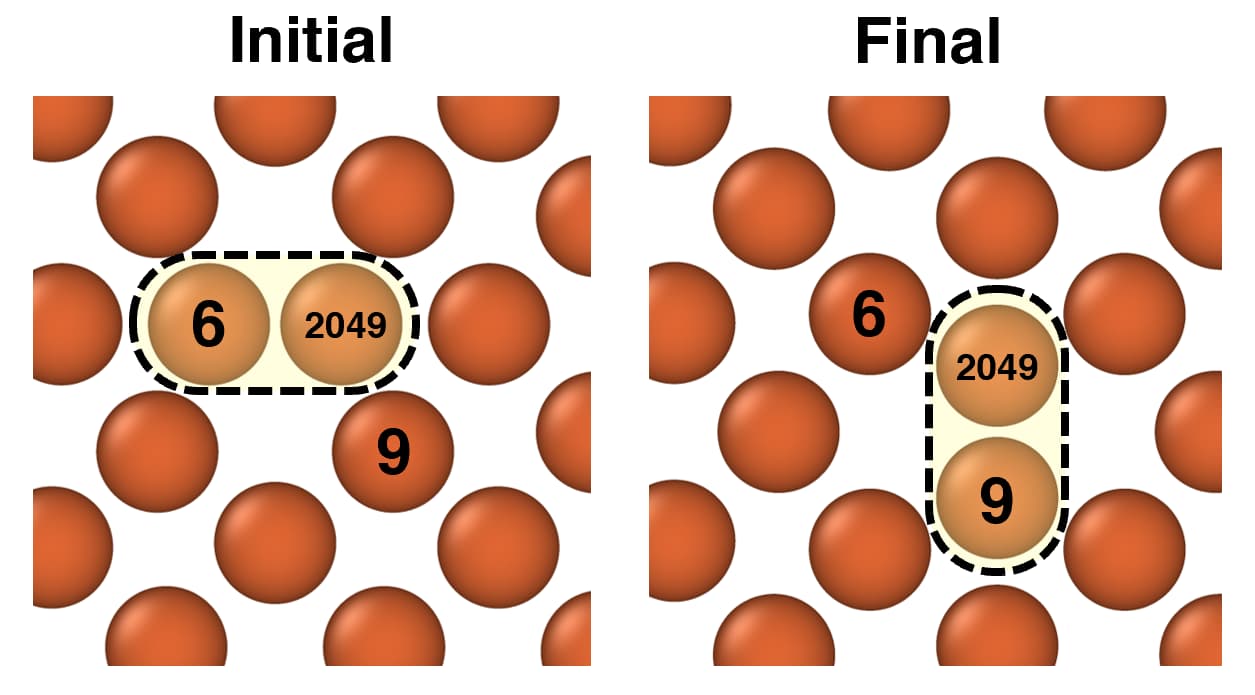
Then I am running NEB simulation with 12 partitions. I am using the following commands:
minimize 1e-15 1e-15 5000 5000
fix 1 all neb 1
thermo 100
timestep 0.01
min_style quickmin
neb 0.0 0.005 100 100 10 final final.txt
I am only taking the co-ordinates of the newly created interstitial Ni atom. The final.txt file -
1
2049 7.04 1.08236 28.16
I am using a 2048 atom system.
But I am getting near zero energy barriers (~3x10^-5 eV). This is the last line of the neb log file:
Step MaxReplicaForce MaxAtomForce GradV0 GradV1 GradVc EBF EBR RDT RD1 PE1 RD2 PE2 RD3 PE3 RD4 PE4 RD5 PE5 RD6 PE6 RD7 PE7 RD8 PE8 RD9 PE9 RD10 PE10 RD11 PE11 RD12 PE12
223 0.0091862862 1.4465175e-05 5.6877652e-09 0.0052672766 0.0077208736 6.0790999e-05 3.1616739e-05 0.053057438 0 -9109.3035 0.31256984 -9109.3035 0.56451477 -9109.3035 0.68781715 -9109.3035 0.74024736 -9109.3035 0.76448997 -9109.3035 0.77446701 -9109.3035 0.77662467 -9109.3035 0.79635679 -9109.3035 0.84244947 -9109.3035 0.91348897 -9109.3035 1 -9109.3035
However, a literature that uses the same potential file that I am using, suggests that I should be getting an energy barrier of about 0.17 eV. Dumbbell rotation-induced interstitial migration energy of pure Ni is found to range from 0.1 to 0.2 eV in other literatures as well.
I have tried creating dumbbells in with different atoms and rotating it with their neighboring atoms. I have also tried making the initial and final configurations with out minimizing at the end, but it didn’t make any difference. Any assistance in solving this issue would be greatly appreciated.
Thanks,
Tawseef