Hi
In my simulation I apply pressure on a liquid as you can see in the picture below. The liquid is placed between two plates. One of them is fixed and another one applies pressure.
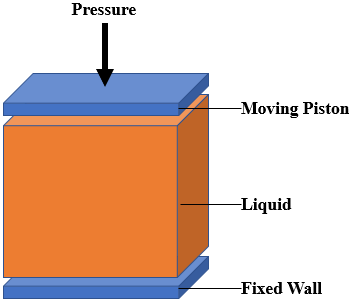
I decided to calculate pressure and temperature by a dump file outputs. I saved x y z vx vy vz fx fy fz. in the dump file.
I calculate temperature by KE = 3/2 N.K_B.T and the result was the thing was expected. For pressure I used P= N.K_B.T/V + 1/3V sum(r_i . f_i) and I set V = the volume of the region, T = the temperature calculated in previous section and for r_i and f_i I used the data saved in the dump file. But when I do this calculation I give wrong answer. My pressure must be 100atm but I see 168 atm. Is it possible for someone to help me?
I run my code parallel, real units is used and the boundaries are periodic in both x and y.
Also you can see the python code I use to calculate pressure.
def calc_pressure(T,cordinates,forces,volume,index,n):
kb = 1.38e-23 # [J/K]
SelectedF = forces[index[:][0],:] * 6.9477e-11
# * 6.9477e-11 to convert kcal/mol.Angestrom to N
SelectedC = cordinates[index[:][0],:] * 1e-10
# * 1e-10 to convert Angestrom to m
P = (n*kb*T)/volume
temp = 0
for i in range(n):
temp = temp + np.dot(SelectedF[i,:],SelectedC[i,:])
P = (P + 1/(volume*3)*temp)*9.8692e-6
# P = N*KB*T/V + 1/(V*3)*sum(ri.fi) and *9.8692e-6 for converting [Pa] to [atm]
return P
1 Like
Computing pressure from F dot r cannot give correct answers with a periodic system if done after the fact. For every pair of atoms that straddles periodic boundaries you have to use the position that was used for the force computation, including those of the periodic images. In LAMMPS the force from such pairs is stored on the corresponding ghost atoms and when computing F dot r, the position of those ghost atoms is used. After that LAMMPS does a so-called reverse communication and sums the forces from the ghost atoms to their canonical local atoms. From then on F dot r cannot give a correct result, but this is the data you are using in your calculation.
Thanks Axel for your response.
So, according to the point you’ve mentioned, what can I do to calculate the pressure?
You can use the pressure computed by LAMMPS.
because of the details of my simulation box, I don’t think that’s possible.
after reaching equilibrium in my system, I remove the moving piston and the wall below it. therefore some liquid will evaporate and I have both vapor and liquid in my system. I want to calculate the pressure of both states and the thing is that my liquid is moving in the box, so it’s not possible to define a fixed region and then calculate their pressure.
In that case, you could let LAMMPS collect the per-atom stress tensor information and dump it with the positions and then compute the pressure within subsets of your system from that. Please see: compute stress/atom command — LAMMPS documentation
One problem with this is the question whether there is a well defined pressure for those components that you can compute under those circumstances in the first place.
Thanks Axel. 
It seems the pressure can be computed in this way correctly.