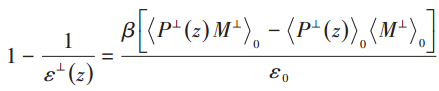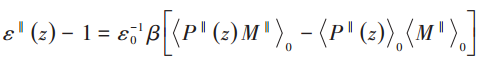I am trying to calculate the dielectric constant at different distances within a graphene channel using lammps. According to the formulae in the literature, I am using the compute chunk/atom bin/1d command to divide the graphene channel into chunks at different distances and calculate the electric dipole moments therein in order to compute the local polarisation density P(r).
The dielectric constant calculated in the literature at different distances is of the same order of magnitude as the bulk dielectric constant, but my calculated dielectric constant is two orders of magnitude larger than the one calculated in the article. I tried subtracting the product of electrostatic charge and centre of mass inside the chunk, resetting the value of image, and using unwrapped coordinates when calculating the electric dipole moment, but the results still change very little.
Do I have any problems with the way the physical quantities are calculated in the formulae, or is it the use of the lammps command?
Sincerely looking for your advice and help
Thank you
Best regards
Your attached input script is extremely complex with many commands, some apparently trying to do the same thing in slightly different ways, and many variables whose names do not obviously state what is measured. If we can’t understand what your script is trying to do, it is very difficult for us to help you
Please state which paper (or papers) your attached screenshots are from. Otherwise it is impossible for us to understand the wider context of these formulae (for example what “M” is).
Please state what the literature values are or what your value is, and please state whether you were trying to perform an exact replication (which is what we always recommend as a first step) or trying something quite different (in which case it will be even harder for us to work out what’s going on).
Finally, both polarisation and polarisation density are inherently tricky to calculate for all kinds of reasons. In this particular case, hydrogen and oxygen atoms do not have electric dipoles; the water molecules, which do have dipoles, do not have defined “positions” (being multiparticle “groups”). Furthermore, the highly ordered layers of water near graphene make it very difficult to define any continuous function of position. This makes it all the more important for us to know exactly what your literature comparison point is.
Another thing I should add is that since I usually use SPC/E water molecules more often, I started with SPC/E water molecules in my system to try to get close to the results of TIP4P-2005 water molecules in the literature, but the results were more different than I expected.
I really appreciate your reply and apologise for my immature question.
According to the equations in the literature (where M is the electric dipole moment; P(r) is the polarisation density and M = ∫P(r)dv; β=1/kT, where k is Boltzmann’s constant, T is the temperature of the system; ε0 is the vacuum dielectric constant; <> means a systematic averaging of physical quantities therein) the total electric dipole moment and polarisation density need to be calculated, and I tried to indirectly calculate the polarisation density by calculating the electric I tried to calculate the polarisation density indirectly by calculating the electric dipole moment in each chunk. Boundaries were set using bin/1d in the calculation of the electric dipole moments to remove boundary effects, as well as removing the effect of net charge. Since the output of reduce/chunk is a vector, I set up a loop to calculate the polarisation density for each chunk and output the dielectric constant calculation for each chunk one by one.
I’m trying to accurately duplicate the simple case of zero graphene oxide hydroxyl concentration from others’ literature. But in setting up the CHARMM27 force field I simply set the force on the c atom of graphene to zero and only set up the lj interactions between non-bonded atoms. About expressing the average dielectric constant by calculating the root sign of the product of the parallel and perpendicular dielectric constants is a method I saw in other forums, which I tried to calculate in passing, but the result is still not satisfactory.
I apologise that my server calculations have been overwritten by my subsequent misguided attempts, but I can clearly remember my results differing wildly from the literature, to the 7-9th power of 10 or even negative.
The following provides the lammps commands after I added the comments and the article I chose to try to duplicate
Journal of Molecular Liquids 324 (2021) 115139 in.dipole (5.7 KB)
Please invest time in finding a local mentor who can give you detailed, hands-on advice. This will help you get help faster than relying on Internet forums
Wherever possible, it is useful to go as far back to obtain the methodology directly from the source if possible. The J Mol Liquids paper that you attached does not clearly described its methodology (and may in fact be just wrong). That paper does cite its source as Bonthuis’s paper from 2011: https://journals.aps.org/prl/abstract/10.1103/PhysRevLett.107.166102 which uses entirely different methodologies for calculating the polarisation density function:
the transverse polarisation density at z is calculated as the integral of 2D charge density from 0 to z;
the parallel polarisation density at z is apparently calculated as the integrated dipole moment of a thin slab about z divided by the surface area.
You will have to investigate these calculations for yourself.
If you are accidentally overwriting trajectories that’s not a good sign. In general, when you are trying to construct such a complicated analysis procedure, you should always create a simulation trajectory first that you keep a backup of and then separately analyse the trajectory, which you can do in LAMMPS by first using a dump to store the trajectory during simulation and then rerun to test the analysis later.
While it’s still not very clear what the analysis variables in your script are, I noticed you use ids once chunk once in your 1D chunk computes. That’s almost certainly wrong. Any attempt to measure a profile at some co-ordinate z always depends on the atoms at z at that point in time in the simulation, not on atoms which were initially at that z (but will then have diffused away).
Thank you very much for the advice and some of the usability tips, it has been very profitable. It has guided me on how to find the right way to solve the problem. I really appreciate your patience in answering my questions.