Dear community, I would really appreciate your help in solving the problem with calculation freezing when moving from ‘boundary p p p’ to ‘boundary p p s’.
I study the impact of N2 molecule on diamond using many-body (Tersoff type) and ReaxFF pair potentials.
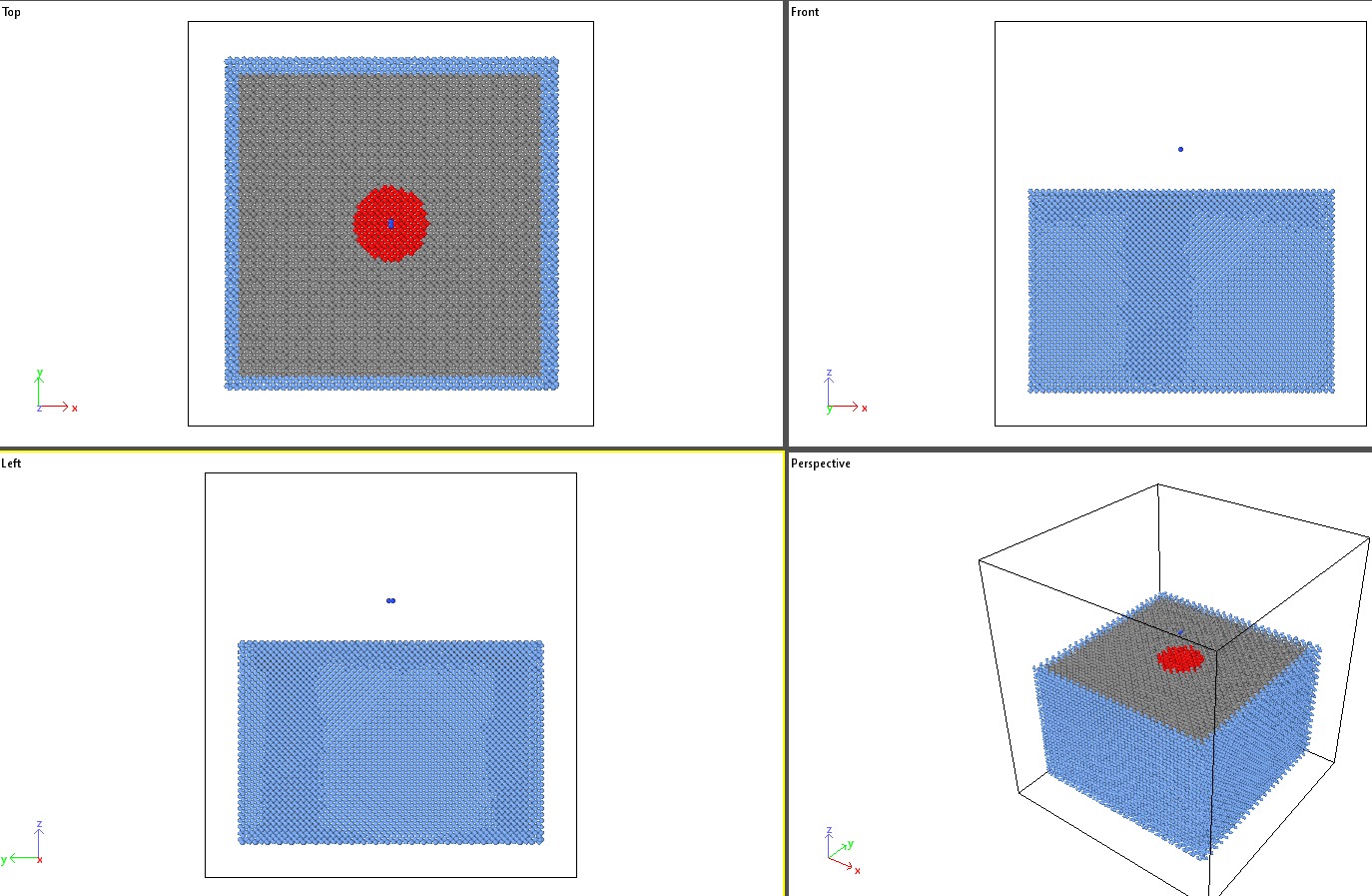
The diamond target is divided into middle and edge groups, where the edge group (shown as blue atoms in the figure) is used to prevent vibrational energy from being reflected from the edges.
First, periodic (p p p) boundary conditions is used. At that conditions minimization of the energy, then NPT, and NVT equilibration are performed. Then boundary is changed to p p s, which is necessary for bombarding the surface in the z direction.
If I use the Tersoff type many-body potential, everything works fine. As soon as I switch to ReaxFF, the computation freezes when going from the p p p boundary to the p p s boundary.
This means that the task keeps running forever without any computation results. Please see the attached input and output files for more details. Note, I tried to change the fix group from ‘middle’ to ‘all’ but it did not help.
Following Axcel’s (to Fernanda_S_Teixeira advice from May 2017) I changed from “boundary p p s” to “boundary p p m”. Thus, everything works fine. I just want to remind that there are no any problems when using the Tersoff type potential.
There are several questions that I would very please for your assistance. What probably I do wrong when use “boundary p p s” and what could be a principial difference when switch to “boundary p p m” so the tasks run with no freezing.
I don’t see this in the LAMMPS manual. What am I missing? In the LAMMPS manual I could see that not only “f” or “m” can be used, but also their combination “fm”. Unfortunately, I don’t understand the physical meaning of the combination “fm”. Thus, could I probably use “sm” or “sf”? But I’d rather understand the combinations before using them.
Thank you and Kind regards,
Victor
N2dia.in (6.8 KB)
N2dia.out (17.1 KB)
N2.dat (198 Bytes)
lmp_control (1.1 KB)
log.N2dia (11.1 KB)
ffield.reax.chon2019 (8.6 KB)