Dear LAMMPSer:
I am curious about how LAMMPS compute the inertia tensor of a molecule composed of finite size spheres.
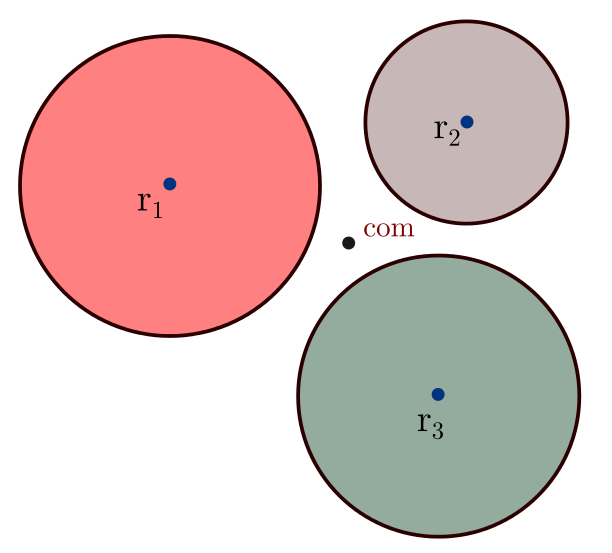
According to the function compute_inertia() in “molecule.cpp”:
…
- if (!inertiaflag) {
- inertiaflag = 1;
- atom->check_mass(FLERR);
- double onemass,dx,dy,dz;
- for (int i = 0; i < 6; i++) itensor[i] = 0.0;
- for (int i = 0; i < natoms; i++) {
- if (rmassflag) onemass = rmass[i];
- else onemass = atom->mass[type[i]];
- dx = dxcom[i][0];
- dy = dxcom[i][1];
- dz = dxcom[i][2];
- itensor[0] += onemass * (dydy + dzdz);
- itensor[1] += onemass * (dxdx + dzdz);
- itensor[2] += onemass * (dxdx + dydy);
- itensor[3] -= onemass * dy*dz;
- itensor[4] -= onemass * dx*dz;
- itensor[5] -= onemass * dx*dy;
- }
- if (radiusflag) {
- for (int i = 0; i < natoms; i++) {
- if (rmassflag) onemass = rmass[i];
- else onemass = atom->mass[type[i]];
- itensor[0] += SINERTIA*onemass * radius[i]*radius[i];
- itensor[1] += SINERTIA*onemass * radius[i]*radius[i];
- itensor[2] += SINERTIA*onemass * radius[i]*radius[i];
- }
- }
- }
…
If the molecule consists of a set of point spheres, then the inertia tensor is simply calculated by the line 6-18.
If the component spheres are finite size (radiusflag is true), then Ixx, Iyy, Izz are updated by adding every sphere’s inertia 2/5* m_i* r_i* r_i via line 23-25.
Can someone give some source about how to calculate the inertia about the center of mass, for a system of non-overlap solid spheres?
Best,