I want to calculate the energy with which spring is pulling a group of atoms in my system. I want to study the peeling mechanism basically. I am attaching the code that I am using. I am stuck as what commands should I give in order to get the necessary information i.e., pulling energy w.r.t coordinates of Centre of mass.
# initialisation
units real
dimension 3
atom_style full
bond_style harmonic
angle_style harmonic
pair_style lj/cut/coul/long 11
kspace_style pppm 1.0e-4
dihedral_style opls
improper_style harmonic
variable pre equal 1
variable tem equal 300
# system definition
read_data data.min.lammps
include PARM.lammps
# simulation setting
group gcar type 1
group gbike type 2
variable zmax equal bound(gcar,zmax)-3.35
variable zmin equal bound(gcar,zmin)+3.35
region rtop block INF INF INF INF ${zmax} INF
region rbot block INF INF INF INF INF ${zmin}
group gtop region rtop
group gbot region rbot
variable xmax equal bound(gtop,xmax)
variable xmin equal bound(gtop,xmin)+90
region rdes block ${xmin} INF INF INF ${zmax} INF
group gdes region rdes
group test id 10965 11002 11003 10620 10619 10581
fix mysf2 gbot setforce 0 NULL 0
velocity gbot set 0 NULL 0
fix pull test smd cvel 5.0 0.00001 tether 27.664753887632 72.85058174439 6.9654 48.770636
fix tether gdes spring/self 10.0
neigh_modify every 1 delay 0 check yes
dump mydmp all atom 1000 dump.lammpstrj
# run
#thermo 10
#minimize 1.0e-6 1.0e-6 1000 10000
fix 4 all nvt temp 300.0 300.0 0.01
timestep 0.01 # (fs)
thermo 1000
run 5000000
Please check the documentation of fix smd carefully. It provides multiple output properties that can be added to the thermo output or written out with fix print or similar. This includes the pulling force. Integrating this force will give you the (free) energy (minus a constant) for the pull across the pull distance. In addition this is also numerically integrated internally and output as the so-called potential of mean force (PMF).
Please note that fix smd is a very old and minimal implementation. Software packages like colvars and Plumed offer much more flexible and sophisticated ways to determine such (free) energies. Both are supported by LAMMPS.
Dear Axel and Giacomo,
I am trying to study the energy expense in the peeling and shearing of graphene flake. For this purpose I am having following problems. I would be grateful if you people can give any suggestions in getting rid of these problems.
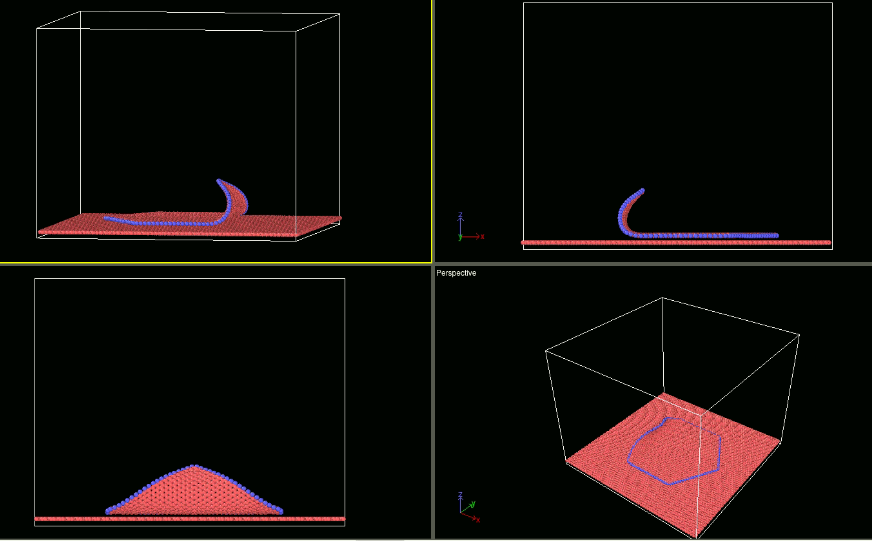
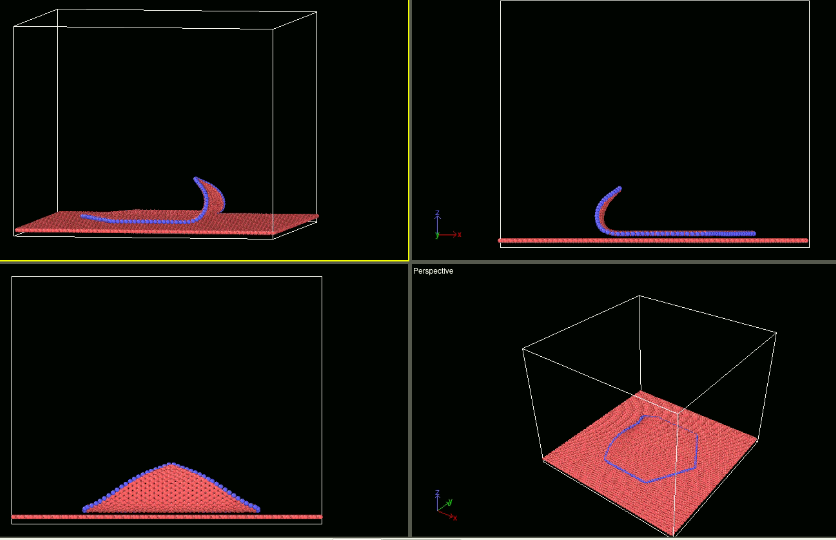
As I have mentioned earlier I am using fix smd for pulling the flake (which I am able to do) for studying the peeling behaviour. For studying the shearing behaviour I intend to use the fix smd too. In that quest I am just changing the direction of velocity (making it negative). However it is not showing any different behaviour than when the velocity is positive. Please guide me what I should do differently to achieve my goal. I am attaching the input scripts and snapshots of my visualisation of trajectory.
a) when the velocity is positive -: input_test1.lammps (1.6 KB) log _14.lammps (882.2 KB)
As suggested by you guys I tried to make use of colvar package. However there is no movement in the flake by using it. I know I might have done something wrong and I tried to figure it out by myself. However, I was unable to rectify my mistakes please let me know my mistake and guide me what I should do next to get the desired result. input_test1.lammps (1.6 KB) PMF_shivi.colvars (391 Bytes) log.lammps (72 KB)
Indeed part of answering why the results of your fix smd and fix colvars computations are different requires knowing the specifics of your system.
One thing you should pay attention to is that you mean to impose the same protocol. Right now, I don’t think you are. I can’t compare reliably the pulling speeds, but in the Colvars setup you added a targetNumStages option with a value of 22.0 (which I’m not sure what it represents).
What that keyword does is making a stationary restraint, which will not move for the entirety of targetNumSteps and then make a discrete move to the next stage. By adding that keyword, you are multiplying by 23 the number of MD steps needed to achieve the same transformation (there are 22 stages plus 1 for the initial one). Please only add keywords that are off by default after checking their documentation.
Ok, it looks the problem is that your pulling group’s center is exactly located on the tether point. That makes the pulling direction ambiguous and subject to numerical noise. You want to place the tether point a good distance away so that fix smd can infer the pull direction vector accurately.
Thank you Axel,
I will change the location of pulling group. I will let you know how it will behave. However, I am still curious how can I impart shearing motion in my flake by fix spring. Although I used fix drag initially for imparting shearing motion in the flake. But I am interested in pushing flake by fix smd.
Best,
Shivam
I tried to change the location of the tether point. In two separate runs, in one run I put the tethering point (100, 100,100 and 50) and in another (100, NULL, NULL, 0.0). However, in both cases flake was stagnant in its initial position and I could not observe any motion as such in it.
I agree with you that the pulling group center was exactly located on the tether point in my previous runs. Then how come the flake was peeled off from its initial position?
Warm Regards,
Shivi input_test1.lammps (1.6 KB) log.lammps (356.7 KB)
Dear Giacomo,
As suggested by you I excluded targetNumStages still the flake is stagnant. I am attaching the log files. input_test1.lammps (1.6 KB) log.lammps (234.1 KB) PMF_shivi.colvars (372 Bytes)
Please look at your thermodynamic output. Doesn’t it strike you as odd that your temperature is so very far from the desired target but then jumps occasionally, sometimes to a very high value?
That suggests that there is something very wrong going on in how you are setting up your simulation, thermalization and time integration.
For example, it doesn’t make sense at all to use fix nvt for all atoms, specifically not the “flake” since that will include the added energy from the pulling. It also is not representing any physical behavior. The exchange of kinetic energy should be just as it happens “naturally”.
Another warning sign is your very small timestep. If your system would be set up properly, the timestep should be at 1fs easily without breaking a sweat.
Since this thread is starting to give the impression that there is something wrong with LAMMPS, I wanted to give a little demonstration that it does work as advertised, so I constructed a very small input example of my own.
A few general remarks. The whole pushing/pulling of the flake and it being somewhat stationary didn’t make sense to me. Based on other simulations (e.g. sticking buckyballs into nanotubes), I would expect very low friction. In fact the flake should diffuse around from the thermal vibrations (its own and the supporting layer. Almost like a hooey machine.
So to have any meaningful pushing/pulling one would have some way to “fuse” the flake with the supporting layer. In this example I emulated a bond by adding a spring between a few atoms of each component. Also I added spring to the center of mass of the supporting layer so it doesn’t float around.
To simplify the procedure (and because I am lazy) I modeled the layers with tersoff (so I didn’t have to create all those bonds across PBC etc). For convenience I also define a few groups that are used to connect the layer to the flake (colored magenta and cyan) and to attach fix smd (gray). There also is the reference point for fix smd (white). Push and pull are the same input, only the sign of the fix smd velocity is changed. Here is the input (note this is not tested to be accurate but simple provided to showcase the functionality itself and it should NOT be used as a template for doing real science).:
units real
atom_style full
special_bonds lj/coul 1.0 1.0 1.0
read_data flake.data extra/atom/types 3
group none empty
change_box none z scale 2.0
pair_style hybrid tersoff tersoff lj/cut 12.0
pair_coeff * * tersoff 1 SiC.tersoff C NULL NULL NULL C
pair_coeff * * tersoff 2 SiC.tersoff NULL C C C NULL
pair_coeff 1 2*4 lj/cut 0.07 3.55
pair_coeff 2*4 5 lj/cut 0.07 3.55
mass * 12.01
group layer type 1
group flake type 2
region pull sphere 28.3 17.2 3.35 3.3
group pull region pull
region upper sphere 20.1 31.6 3.35 3.3
group upper region upper
region lower sphere 20.1 31.6 0.0 3.3
group lower region lower
set group upper type 4
set group pull type 3
set group lower type 5
thermo 100
fix 0 all box/relax x 0.0 y 0.0
fix 1 lower spring couple upper 50.0 0.0 0.0 1.0 3.35
fix a layer spring tether 500.0 25.0 25.0 0.0 0.0
minimize 0.0 0.0 10000 100000
write_dump all atom after_min.lammpstrj
unfix 0
reset_timestep 0
thermo 1000
dump 1 all atom 1000 equilib.lammpstrj
velocity all create 300.0 64653456
fix 2 all npt temp 300.0 300.0 10.0 x 0.0 0.0 1000.0 y 0.0 0.0 1000.0
timestep 1.0
run 100000
undump 1
unfix 2
dump 1 all atom 200 push.lammpstrj
fix 2 pull smd cvel 50.0 -0.0001 tether 20.0 30.0 3.35 0.0
fix 3 flake nve
fix 4 layer nvt temp 300.0 300.0 10.0
run 200000
And here two animations where the gray group is pulled away from the tether point:
and pushed toward the thether
This all looks as expected and one can clearly see that fix smd does what its documentation says it does.