Code :
This is the control script for LAMMPS
echo both
#-------------------------------------------------------------------------------
Stage 2.1: Initialize LAMMPS run
#-------------------------------------------------------------------------------
package omp 4
units real
atom_style atomic
variable T equal 450 # kelvin
variable V equal vol
variable dt equal 2.0
variable p equal 200 # correlation length all in angstrom
variable s equal 10 # sample interval
variable d equal $p*$s # dump interval
convert from LAMMPS real units to SI
variable kB equal 1.3806504e-23 # [J/K] Boltzmann
variable kCal2J equal 4186.0/6.02214e23
variable A2m equal 1.0e-10
variable fs2s equal 1.0e-15
variable convert equal {kCal2J}*{kCal2J}/{fs2s}/{A2m} #4.83e-16
boundary p p p
processors * * *
#create_box 3 box
read_data MOS2.data
#create_box 2 box
replicate 126 36 1
#replicate 526 660 10
group watom type 1
group seatom type 2
neighbor 1.0 bin
#neigh_modify delay 0 every 1 check yes
neigh_modify delay 3
pair_style lj/cut/omp 4.035
pair_coeff 1 1 0.00243 2.719 3.052
pair_coeff 2 2 0.01187 3.595 4.035
pair_coeff 1 2 0.02489 3.157 3.544
timestep ${dt}
thermo $d
equilibration and thermalization
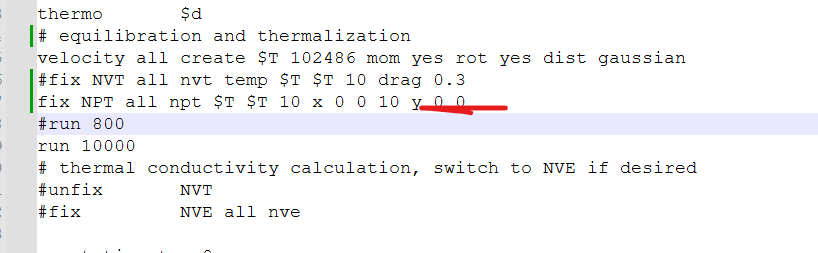
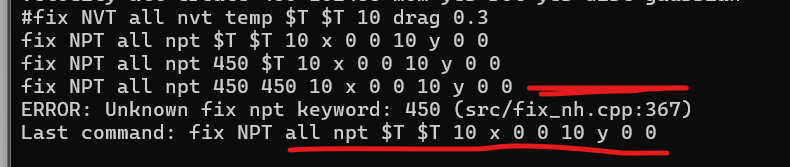
velocity all create $T 102486 mom yes rot yes dist gaussian
fix NVT all nvt temp $T $T 10 drag 0.3
#run 800
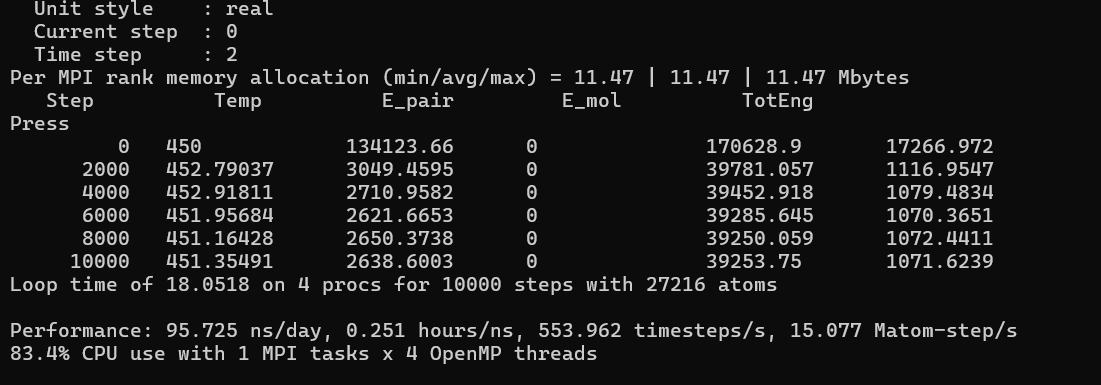
run 10000
thermal conductivity calculation, switch to NVE if desired
#unfix NVT
#fix NVE all nve
reset_timestep 0
compute myKE all ke/atom
compute myPE all pe/atom
compute myStress all stress/atom NULL virial
compute flux all heat/flux myKE myPE myStress
variable Jx equal c_flux[1]/vol
variable Jy equal c_flux[2]/vol
variable Jz equal c_flux[3]/vol
fix JJ all ave/correlate $s $p d &
c_flux[1] c_flux[2] c_flux[3] type auto file J0Jt.dat ave running
variable scale equal {convert}/${kB}/$T/$T/$V*s*{dt}
variable k11 equal trap(f_JJ[3]){scale}
variable k22 equal trap(f_JJ[4])*{scale}
variable k33 equal trap(f_JJ[5])${scale}
thermo_style custom step temp v_Jx v_Jy v_Jz v_k11 v_k22 v_k33
run 6000
variable k equal (v_k11+v_k22+v_k33)/3.0
variable ndens equal count(all)/vol
#print " volume: $vol /m^3 "
print “average conductivity: $k[W/mK] @ T K, {ndens} /A^3” # kappa and density
Data file :
#previous data
LAMMPS 1_3 monolayer
6 atoms
2 atom types
Cell: Hexagonal
0.0000 3.16000 xlo xhi
0.0000 5.47300 ylo yhi
0.0000 32.0000 zlo zhi
Masses
1 95.940 # Mo
2 32.065 # Se1
Atoms
1 1 1.580 4.56107 16.760
2 1 0.000 1.82443 16.760
3 2 1.580 0.91221 18.300
4 2 0.000 3.64885 18.300
5 2 1.580 0.91221 15.220
6 2 0.000 3.64885 15.220
output :