In my simulation, the wall movement is specified by the spatiotemporal data written in a file (either ASCII or binary). I’ve known that there are mainly four ways in LAMMPS to specify the displacement/velocity. displace_atoms and velocity are not applicable because they are executed only once rather than at every time step. Since the velocity of wall movement can change with time, only fix move and fix python/move are applicable. However, fix move cannot read data from a file (one way is to use variable atomfile and variable delete at every timestep but this is too awkward and I need to prepare many data files).
Another way is to use fix python/move, where I can read data use Python’s reader. The only challenge is that fix python/move can only operate on all the atoms but I’m not sure. Examples in example/python/py_nve.py have assert(self.group_name == "all"). But in my simulation, I only want to specify the wall movement but not the fluid therein. The fluid movement is integrated by fix rheo. The cpp source checks whether an atom belongs to a group by mask[i] & groupbit, but how can this be done in Python lammps module? If it is not feasible, is there any other convenient way to implement this idea?
Thanks in advance!
==========================================
Whoops, I just forgot that one can specify the group in the Python function lmp.extract_variable. Maybe I can use this:
def __init__(self, ptr, group_name="all"):
super(NVE, self).__init__(ptr)
def initial_integrate(self, vflag):
x = self.lmp.numpy.extract_atom("x", group=self.group_name)
v = self.lmp.numpy.extract_atom("v", group=self.group_name)
for i in range(x.shape[0]):
...
Now here is another question: how can I debug this python code?
Sorry, I confused extract_atoms with extract_variables. extract_atoms does not receive the group argument. I just test the method you provided in the example script, it works.
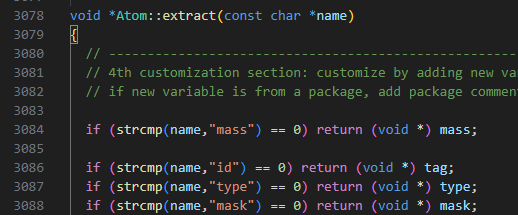
Another question is how to obtain the atom tags. I’m going to use this command in the parallel mode. So I need to map the wall movement which is stored by global atom-IDs to atoms of each processor with local atom-IDs. In the callback function of fix external, tags could be easily obtained. But tags is not an argument here. I try to use
tag = lmp.numpy.extract_atoms('tag')
But I got None. Now that I can get mask, x, f, etc., by extract_atom, why can’t I get tag by the same method?
=====================
After checking the source code, I got the answer: I should use 'id' rather than tag to for atom tags extraction, which lets me think of what we specify in the dump command is id instead of tag. But I suggest this should be mentioned as a special case in the documentation of extract_atoms.
@jasmine969 Please note that my example only works when you do not delete groups. The list of groups is special, since it can have “holes” in them (unlike the list of variables, or computes, or fixes or dumps etc.). Thus the code in the LAMMPS library interface and particularly the LAMMPS python module needs to create a special case when querying groups so that it always returns a list of all 32 possible groups and then the unused groups have the value “None” in that list. Without these changes, LAMMPS will either crash with a segfault or produce incorrect group bitflags.
Corresponding changes are in a pending pull requests and have also been backported to the stable version.