Hello Sir/Ms,
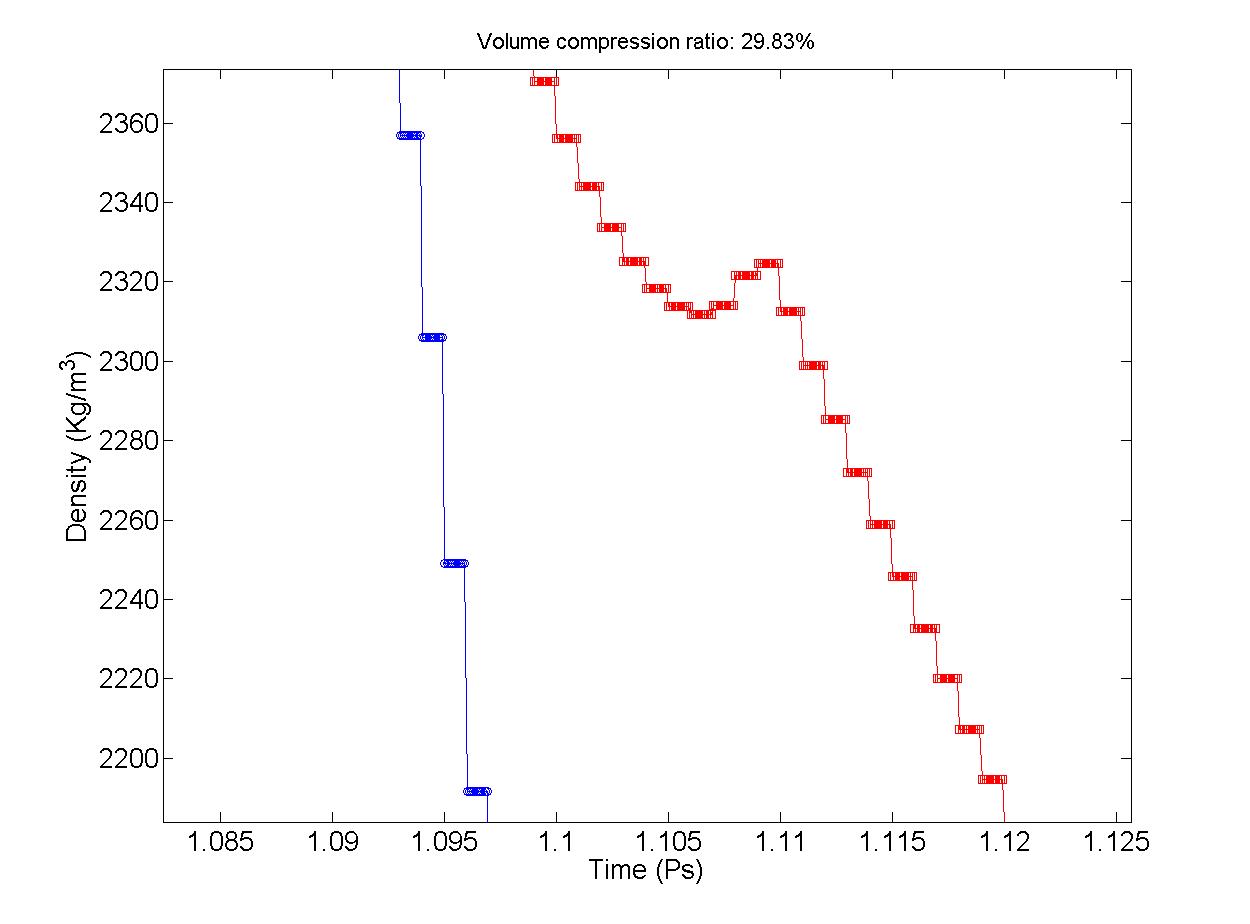
I am using LAMMPS with ReaxFF to study the detonation of explosive (HMX). After simulation, I got the time-density (time-volume) curve like the attached pic. If you zoom the curve, you can see for each piece of time, the density will remain the same (I counted the dots on each piece of horizontal line segment, they have the same number 10). So I guess there must be some parameter in LAMMPS algorithm to determine this number. Now I wonder if I can modify this parameter? What influence it will have if I modify the parameter? Will the accuracy be reduced or what? Thanks for your reply.
Guangyu Wang
What command did you use to change the volume?
Ray
Hello Sir/Ms,
I am using LAMMPS with ReaxFF to study the detonation of explosive (HMX).
After simulation, I got the time-density (time-volume) curve like the
attached pic. If you zoom the curve, you can see for each piece of time, the
density will remain the same (I counted the dots on each piece of horizontal
line segment, they have the same number 10). So I guess there must be some
parameter in LAMMPS algorithm to determine this number. Now I wonder if I
can modify this parameter? What influence it will have if I modify the
parameter? Will the accuracy be reduced or what? Thanks for your reply.
there is not enough information in your description to give a
meaningful suggestion.
and in particular, what kind of "algorithm" are you referring to?
axel.
Ray,
I used “fix 1 all nve” in this simulation. Since I modified the boundary from p to s so the volume can change automatically.
Guangyu Wang
If that is the case, then the volume change has to result from other commands you used in the script.
It is not clear what is it that you wanted to do: minimization at 0K? relaxation at some finite temperature? shock loading?
Ray
If that is the case, then the volume change has to result from other
commands you used in the script.
shrinkwrap boundaries are only updated during neighbor list updates, IIRC.
It is not clear what is it that you wanted to do: minimization at 0K?
relaxation at some finite temperature? shock loading?
it appears the simulation has a fundamental problem, too. inferring
the probe volume from the shrinkwrap box extent can lead to a very
large error if a single atom leaves the slab. not to mention that
switching from a bulk system to a thin slab (this is what switching
from p to s boundaries implies) requires a reequilibration and is
ultimately the investigation of a quite different setup.
axel.
Ray,
In fact I want to simulate the detonation of HMX. First, I set the boundary for X,Y,Z to be periodic. Then I use NPHUG (NPT) to compress the volume of HMX to 70% of its original volume. Then I will maintain the state of compressed HMX for a while using NVE. Then I will modify the boundary from periodic to, for example, s (shrink) p p, and at the same time, I use NVE ensemble and let the system expand adiabatically.
Guangyu Wang
Axel,
I need to simulate the isentropic expansion of reactive system after detonation. That is why I modify the boundary from p to s. At the same time, I use NVE to keep the energy of the system constant.
Guangyu Wang
Axel,
The expansion of reactive system itself is non-equalibrium process. Do you think I need to reequilibrate the system during the expansion process? That will be quite different from reality.
Guangyu Wang
Axel,
I need to simulate the isentropic expansion of reactive system after
detonation. That is why I modify the boundary from p to s. At the same time,
that is a rather unusual setup. i am not an expert in these kind of
simulations. i cannot give any recommendation for that.
I use NVE to keep the energy of the system constant.
fix nve does *not* "keep the energy constant". all it does is plain
velocity verlet time integration. for a simple bulk system with no
other fixes, this will result in an nve ensemble. nothing more,
nothing less.
Everything is reasonable until you changed the boundary conditions from periodic to non-periodic (shrinkwrapped). By doing so, you introduce two newly created surfaces that are not equilibrated and worst of all, with lots of dangling bonds. If you want to create surfaces, then the surfaces must be created and equilibrated before compression.
Ray
Ray,
The expansion of detonation product is a dynamic non-equilibrium process from compressed state. I need to obtain the density-pressure curve during this process. What do you suggest for the simulation of this process? Thanks.
Guangyu
You might be able to keep the periodic boundary and use a combination of nve + press/berendsen to relax to zero pressure.
Or you can use fix msst from the first place and unfix it when you want to observe free expansion.
In any case, you can not create new surfaces like you did previously.
Ray
Ray
I tried the way (nve+fix press/berendsen) you told but I found it is hard to determine the Pdamp. Lammps will always crash at the first few steps. I came up with another method: NVE+fix deform to manually control the expansion of the system. Do you think it is reasonable? Thanks.
Guangyu
Ray
I tried the way (nve+fix press/berendsen) you told but I found it is hard to determine the Pdamp. Lammps will always crash at the first few steps.
Are you allowing only one direction to relax? Pdamp is typically 1000 time units. If it crashes within a few steps, something bad with your model is going on.
I came up with another method: NVE+fix deform to manually control the expansion of the system. Do you think it is reasonable? Thanks.
Are you sure it is still adiabatic expansion by doing so?
Ray
Ray,
I need to test two release methods: 1st is to release in only one direction, 2nd is to release in 3 directions. During the release, the system will do work to the outer environment.
Guangyu
Ray,
Can I use fix nphug to release the system? Thanks.
Guangyu
I am not sure. You will have to test it and verify that results make sense.
Ray
Ray,
The result is not good. But when I use modified boundary from p to s+nve to simulate the free expansion of the system. The simulation does agrees well with experiment. I don’t know why.
Guangyu Wang
By changing the boundary from p to s you are creating new surfaces - which is definitely not correct.
The agreement is either by chance or the surface effect is small, but I wound’t be satisfied with results (good or bad) from bad setup.
Ray