Hi everyone , I’m new to LAMMPS. I have this system of graphene/PEO and I would like to study its global and local mechanical properties and so compute strains and stresses. After cooling the system, I want to re orient graphene so that it is parallel to x-axis to ease the evaluation of mechanical properties. After centering the system, I tried rotating it using a code with Python but I had a problem that it didn’t respect the periodic boundary conditions that I have on the box. How can I solve this problem?
And is it possible if I leave graphene inclined to define a block region that is parallel to graphene with the same inclination for the purpose of computing stress per atom in this region that is the interphase between the polymer and graphene atoms?
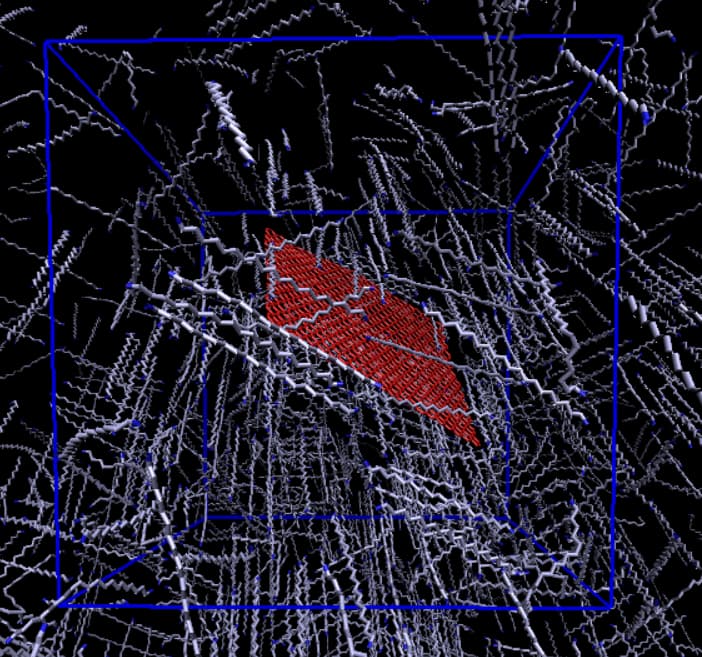
Your image looks strange as if the equilibration didn’t really work and rather your polymers are still in an unphysical stretched geometry.
What you are asking for cannot easily be done because you have a bulk system with periodic boundary conditions. Any rotation outside of 180 rotations are impossible.
What you should have done is to either immobilize your graphene sheet from the beginning and equilibrate only the remaining system first, or apply a position restrain to several points of the sheet, so it can relax but not rotate significantly. Or a combination of the first step, followed by the second.
Since you are new to LAMMPS, an additional recommendation. Please start your LAMMPS simulations with a simpler, single component system and get some practice with those first. The classic sequence would be "LJ liquid Argon, TIP3P or SPC or similar liquid water, a single component polymer melt and only after you have managed to run successful simulations of those reproducing data and results reported in suitable publications describing simulations of those materials should you start with your simulation project. Anything else will lead to significant confusion and problems because you are lacking crucial experience. If at all possible, you should obtain assistance from a local person that can tutor you (on MD simulations in general is probably more needed than LAMMPS specifically. This specific LAMMPS questions, we can help with, the general MD advice is beyond the scope of the LAMMPS forum categories).
Thank you for your reply.
The image resembles crystallization of the polymer chains during cooling.
The graphene sheet was actually inclined from the beginning before equilibration happened. Is it possible to enlrage the simulation box , apply the rotation in Python , then shrink it back without violating the periodic boundary conditions?
Can you answer my question about the block region that matches the inclination of graphene sheet?
No. You are missing the key point of periodic boundary conditions, i.e. that the system continues as if there were no boundaries. However, that conflicts with arbitrary rotations.
Please check the documentation. There are “move” and “rotate” keywords that might do what you are asking for, but I have never used them, so I cannot say for certain. As with all functionality in a software that is new to you, you are advised to construct small test systems/cases and validate all functionality independently and incrementally. That is just common sense for any software not only LAMMPS.