Dear all,
I’m new to using Moltemplate. I’m trying to prepare the data file of a structure containing 50 LiPF6s and packaged with Packmol. (I don’t want the Lithium bonded to the Fluorine.) Firstly, I completed the Packmol part by preparing the pack.in file as follows.
tolerance 2.0
filetype pdb
output system.pdb
add_box_sides 0.0
structure new.pdb
number 50
inside box 0.0 0.0 0.0 15.0 15.0 15.0
end structure
I continued as ethylene+benzene_Packmol file in the moltemplate folder. So, for LiPF6 I wrote the information I read in the oplsaa.lt file in to the LiPF6.lt file as follows.
import “oplsaa.lt”
LiPF6 inherits OPLSAA {
atomID molID atomType charge coordX coordY coordZ
write(‘Data Atoms’) {
$atom:P $mol @atom:726 0.00 -1.051 1.315 -0.000
$atom:F1 $mol @atom:727 0.00 -0.314 2.271 1.222
$atom:F2 $mol @atom:727 0.00 -1.788 0.359 -1.222
$atom:F3 $mol @atom:727 0.00 0.323 0.283 -0.021
$atom:F4 $mol @atom:727 0.00 -0.328 2.301 -1.207
$atom:F5 $mol @atom:727 0.00 -1.773 0.328 1.207
$atom:F6 $mol @atom:727 0.00 -2.424 2.346 0.021
$atom:Li $mol @atom:879 0.00 3.044 0.685 -1.952
}
A list of the bonds in the molecule:
BondID AtomID1 AtomID2
write(‘Data Bond List’) {
$bond:PF1 $atom:P $atom:F1
$bond:PF2 $atom:P $atom:F2
$bond:PF3 $atom:P $atom:F3
$bond:PF4 $atom:P $atom:F4
$bond:PF5 $atom:P $atom:F5
$bond:PF6 $atom:P $atom:F6
}
}
Finally, I prepared system.lt file as follows.
Import the force field.
import /home/cinar/bin/moltemplate/moltemplate/force_fields/oplsaa.lt
import LiPF6.lt # ← defines the “LiPF6” molecule type.
Set the simulation box.
write_once(“Data Boundary”) {
0.0 15.00 xlo xhi
0.0 15.00 ylo yhi
0.0 15.00 zlo zhi
}
Create 50 “LiPF6” molecules
system = new LiPF6[50]
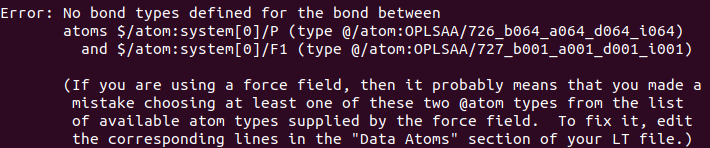
And I ran it with moltemplate.sh -pdb system.pdb system.lt command. But I’m getting this error.
Actually, I think I chose the atom types correctly. Please, how can this be resolved? Sorry for the long post. I really appreciate your help.
Hi @icinar2,
This is a question solely related to moltemplate. You might find more insightful information on the moltemplate website where you can also get troubleshooting, examples as well as the contact info of Andrew Jewett who wrote and maintain the code.
First of all, please read the forum guidelines and do some research before posting questions.
As @Germain suggested, this issue is really a missing parameter in the OPLS-AA force distributed with Moltemplate, so please raise an issue on GitHub. Also, please read this discussion about the versioning of the OPLS-AA force field.
The error explained: the bond coefficient for the hexafluorophosphate ion is not defined in the oplsaa.lt file. You can fix this by adding the relevant command to the definition of LiPF6.lt:
# No need to define the absolute path if you configured Moltemplate correctly ;)
import oplsaa.lt
LiPF6 inherits OPLSAA {
# atomID molID atomType charge coordX coordY coordZ
write("Data Atoms") {
$atom:P $mol @atom:726 0.00 -1.051 1.315 -0.000
$atom:F1 $mol @atom:727 0.00 -0.314 2.271 1.222
$atom:F2 $mol @atom:727 0.00 -1.788 0.359 -1.222
$atom:F3 $mol @atom:727 0.00 0.323 0.283 -0.021
$atom:F4 $mol @atom:727 0.00 -0.328 2.301 -1.207
$atom:F5 $mol @atom:727 0.00 -1.773 0.328 1.207
$atom:F6 $mol @atom:727 0.00 -2.424 2.346 0.021
$atom:Li $mol @atom:879 0.00 3.044 0.685 -1.952
}
# A list of the bonds in the molecule:
# BondID AtomID1 AtomID2
write("Data Bond List") {
$bond:PF1 $atom:P $atom:F1
$bond:PF2 $atom:P $atom:F2
$bond:PF3 $atom:P $atom:F3
$bond:PF4 $atom:P $atom:F4
$bond:PF5 $atom:P $atom:F5
$bond:PF6 $atom:P $atom:F6
}
# Add the missing coefficients and types:
write_once("In Settings") {
bond_coeff @bond:064_001 370.46 1.606
angle_coeff @angle:001_064_001 139.22 90.0
}
write_once("Data Bonds By Type") {
@bond:064_001 @atom:*_b001*_a*_d*_i* @atom:*_b064*_a*_d*_i*
}
write_once("Data Angles By Type") {
@angle:001_064_001 @atom:*_b*_a001*_d*_i* @atom:*_b*_a064*_d*_i* @atom:*_b*_a001*_d*_i*
}
}
I have taken the bond coefficient from this repository, but you would do to the community a better favour if you could identify the missing coefficient in the original OPLS-AA version. Please note that the OPLS-AA version in Moltemplate has been converted from:
The parameters supplied with Tinker are from “OPLS All-Atom Parameters for Organic Molecules, Ions, Peptides & Nucleic Acids, July 2008” as provided by W. L. Jorgensen, Yale University during June 2009. These parameters are taken from those distributed with BOSS Version 4.8.
PS I haven’t checked this choice of parameters, so please be very careful and compare this force field with some references.
1 Like
I realised that the Data Angles By Type will assign the same coefficient to every F–P–F triplet, which is not correct as you do not want to bend axial F atoms in an octahedral structure. For this one, you must specify the bond and angle lists manually. Maybe this is the reason these coefficients were not included in the original OPLS-AA file, but that’s just a guess.
Revised LiPF6.lt
# No need to define the absolute path
import oplsaa.lt
LiPF6 inherits OPLSAA {
# atomID molID atomType charge coordX coordY coordZ
write("Data Atoms") {
$atom:P $mol @atom:726 0.00 -1.051 1.315 -0.000
$atom:F1 $mol @atom:727 0.00 -0.314 2.271 1.222
$atom:F2 $mol @atom:727 0.00 -1.788 0.359 -1.222
$atom:F3 $mol @atom:727 0.00 0.323 0.283 -0.021
$atom:F4 $mol @atom:727 0.00 -0.328 2.301 -1.207
$atom:F5 $mol @atom:727 0.00 -1.773 0.328 1.207
$atom:F6 $mol @atom:727 0.00 -2.424 2.346 0.021
$atom:Li $mol @atom:879 0.00 3.044 0.685 -1.952
}
# Missing coefficients from doi:10.1021/ct900009a and doi:10.1021/acs.jctc.7b00520
write_once("In Settings") {
bond_coeff @bond:064_001 370.46 1.606
angle_coeff @angle:001_064_001 139.22 90.0
}
# List of bonds and angles.
write("Data Bonds") {
$bond:PF1 @bond:064_001 $atom:P $atom:F1
$bond:PF2 @bond:064_001 $atom:P $atom:F2
$bond:PF3 @bond:064_001 $atom:P $atom:F3
$bond:PF4 @bond:064_001 $atom:P $atom:F4
$bond:PF5 @bond:064_001 $atom:P $atom:F5
$bond:PF6 @bond:064_001 $atom:P $atom:F6
}
write("Data Angles") {
$angle:a1 @angle:001_064_001 $atom:F1 $atom:P $atom:F3
$angle:a2 @angle:001_064_001 $atom:F1 $atom:P $atom:F4
$angle:a3 @angle:001_064_001 $atom:F1 $atom:P $atom:F5
$angle:a4 @angle:001_064_001 $atom:F1 $atom:P $atom:F6
$angle:a5 @angle:001_064_001 $atom:F2 $atom:P $atom:F3
$angle:a6 @angle:001_064_001 $atom:F2 $atom:P $atom:F4
$angle:a7 @angle:001_064_001 $atom:F2 $atom:P $atom:F5
$angle:a8 @angle:001_064_001 $atom:F2 $atom:P $atom:F6
$angle:a9 @angle:001_064_001 $atom:F3 $atom:P $atom:F4
$angle:a10 @angle:001_064_001 $atom:F3 $atom:P $atom:F5
$angle:a11 @angle:001_064_001 $atom:F4 $atom:P $atom:F6
$angle:a12 @angle:001_064_001 $atom:F5 $atom:P $atom:F6
}
}
First of all, thank you for your answers @Germain @hothello . I was able to run the LiPF6.lt file only when I revised it as follows, but this time there is something strange in the data file. There are many atom, bond, angle, dihedral and improper types. I could not get over this problem. I added both files.
I experienced the same situation with the H2O molecule I prepared to test. I think there is something overlooked.
LiPF6.lt (1.5 KB)
system.data (51.9 KB)
There is nothing strange in the data file. OPLSAA contains 906 atom types and you have all of them in the force field specifications. If you want to remove unused atom types, just run the utility cleanup_moltemplate.sh after running moltemplate. Please refer to the manual and the examples provided with the source code.
1 Like